
RNA干扰抑制IL-23表达对感染后内脏高敏感小鼠肠黏膜固有层DC活化Th17细胞功能的影响*
2012-10-22 12:10汪之沫王文峰龙艳芹侯晓华
胃肠病学 2012年2期
汪之沫 王文峰 龙艳芹 汪 欢 钱 伟 侯晓华
华中科技大学同济医学院附属协和医院消化内科(430022)
感染后肠易激综合征(postinfectious irritable bowel syndrome,PI-IBS)系指急性胃肠道感染治愈后,部分患者仍存在肠道动力和感觉异常,出现腹痛、腹泻、便秘等肠道相关症状。研究发现PI-IBS患者肠黏膜固有层T细胞浸润增多,伴炎性细胞因子如白细胞介素-1β(IL-1β)等表达增加[1,2];T 细胞数量减少后,IBS症状随之减轻[3]。本课题组的前期研究[4]通过建立旋毛虫感染后内脏高敏感小鼠模型模拟人类PI-IBS的自然过程,发现模型小鼠肠黏膜固有层树突细胞(DC)诱导活化Th17细胞与肠道感染消退后肠黏膜免疫系统的持续激活有关,源自模型小鼠肠黏膜固有层的DC与脾脏CD4+T细胞共培养能诱导其分化为Th17细胞,使IL-17分泌增多。关于DC活化Th17细胞的具体机制,目前仍不甚清楚,推测可能与DC分泌IL-23有关。本研究应用RNA干扰(RNAi)技术抑制DC分泌IL-23,通过观察经小鼠IL-23小发夹RNA(shRNA)干扰的DC诱导CD4+T细胞分化为Th17细胞功能的变化,探讨感染后内脏高敏感小鼠肠黏膜固有层DC活化Th17细胞的机制,以进一步明确DC在肠道感染消退后肠黏膜免疫系统持续激活中的作用。
材料与方法
一、实验动物分组和造模
雄性SPF级NIH小鼠由武汉生物制品研究所提供,6~8 周龄,体质量(25.0±2.3) g,饲养于华中科技大学同济医学院实验动物学部。旋毛虫幼虫由华中科技大学同济医学院寄生虫学教研室提供。
12只NIH小鼠随机分为两组,每组6只。对照组以0.2 mL 0.9%NaCl溶液灌胃;模型组以0.2 mL含300条旋毛虫幼虫的0.9%NaCl溶液灌胃[5]。8周后处死两组小鼠,取肠道和脾脏进行实验。饲养过程中无动物死亡。
小鼠处死前行结肠气囊扩张腹壁回撤反射(AWR)评分,处死后取空肠、末端回肠、近端结肠、远端结肠标本行组织病理学检查,判断模型组小鼠感染后内脏高敏感模型建立是否成功。具体步骤参照本课题组前期研究[4]。
二、肠黏膜固有层DC和脾脏CD4+T细胞的分离纯化
模型组小鼠处死后浸泡于75%乙醇中3 min,于超净台内取全部空肠、回肠和结肠,PBS充分冲洗,剪成0.5 cm×0.5 cm左右的片状,参照本课题组前期研究[4]步骤获取肠黏膜固有层单个核细胞,以MACS®CD11c 免疫磁珠(Miltenyi Biotec)分选DC,操作按试剂盒说明书进行。所获细胞悬浮于含10%胎牛血清的RPMI1640完全培养基中。流式细胞术检测显示CD11c阳性率为78.85%±10.11%,台盼蓝拒染实验显示细胞活性≥90%。
另取脾脏研碎,参照本课题组前期研究[4]步骤获取单个核细胞,以MACS®CD4免疫磁珠(Miltenyi Biotec)分选CD4+细胞,操作按试剂盒说明书进行。所获细胞悬浮于含10%胎牛血清的RPMI1640完全培养基中。流式细胞术检测显示CD4阳性率为98.40%±1.82%,台盼蓝拒染实验显示细胞活性≥99%。
三、小鼠IL-23 shRNA干扰质粒的构建和鉴定
设计、合成能产生小鼠IL-23 shRNA的两条互补单链DNA模板(A链和B链),A链序列为5′-CACCAGCAGCTCTCTCGGAATCTTTCAAGACG AGATTCCGAGAGAGCTGCT TTTTTT G-3′,B 链序列为 5′-AGCT C AAAAAA AGCAGCTCTCTCGGAATCT CGTCTTGAA AGATTCCGAGAGAGCTGCT-3′,由武汉晶赛生物工程技术有限公司合成。A链与B链互补结合后形成双链DNA模板,结构为CACC+正义序列+发夹环序列+反义序列+终止信号序列+SacⅠ,其中正义序列AGCAGCTCTCTCGGAATCT与小鼠IL-23基因p19亚基的一段序列同源,经BLAST同源序列比对,不与小鼠基因组其他基因序列同源。模板两端引入酶切位点黏端序列以便于与载体连接。同时设计、合成能产生与小鼠IL-23基因无同源序列的无关序列shRNA的两条互补单链DNA模板作为对照。
分别以30 μL退火缓冲液溶解合成的1 OD(1 OD DNA=50 ng/μL DNA)A 链和 B 链基因片段,各取 2 μL,加入 16 μL 退火缓冲液混匀,94 ℃水浴退火,自然冷却至室温。取1 μL退火连接产物,加入99 μL H2O作100倍稀释,-20℃保存备用。
以Eco31Ⅰ酶切线性化pGenesil-1.1质粒表达载体,1%琼脂糖凝胶电泳回收大片段。将上述稀释退火片段与质粒pGenesil-1.1连接,连接反应体系内含稀释退火片段1 μL、质粒pGenesil-1.11 μL、10×连接缓冲液 1 μL、T4 DNA 连接酶 1 μL 和 H2O 6 μL,37℃水浴反应过夜。取5 μL连接产物转化DH5α感受态细胞,涂布于含Kanar抗性(终浓度30 μg/mL)的LB平板上,37℃恒温箱培养过夜。挑取单克隆菌落接种于5 mL含Kanar抗性(终浓度30 μg/mL)的 LB 培养液中,37 ℃恒温摇床(7×g)培养过夜。
以质粒小提试剂盒[天根生化科技(北京)有限公司]小量提取质粒,SacⅠ酶切鉴定,酶切反应体系内含质粒 DNA 8.5 μL、10×酶切缓冲液 1 μL 和SacⅠ 0.5 μL,37 ℃水浴反应 3 h,1%琼脂糖凝胶电泳。如酶切鉴定显示切下预期大小的片段,则送含有转化质粒的菌液测序(华大基因)。如测序正确,以质粒大提试剂盒[天根生化科技(北京)有限公司]大量提取质粒,测定浓度后-20℃保存备用。
四、DC转染实验
实验设置IL-23 shRNA组(A组)、空脂质体组(B组)和无关序列shRNA组(C组),每组设2个复孔。分离纯化得到的肠黏膜固有层DC培养于含10%胎牛血清的RPMI1640完全培养基中,接种于6孔板,待细胞汇合率达90%时,以LipofectamineTM2000转染试剂(InvitrogenTM,Life Technologies Corporation)转染相应干扰质粒脂质体复合物或空脂质体,操作按试剂盒说明书进行。转染后24 h收获细胞,以细胞计数板计数后备用。收集各组DC转染前和转染后24 h培养上清液,以备检测IL-23水平。此步骤重复3次。
五、转染后DC与CD4+T细胞共培养
A、B、C 三组分别计数 5×105个 DC,与 1.5×106个CD4+T细胞接种于12孔板,以含10%胎牛血清的RPMI1640完全培养基定容至1 mL,另设置CD4+T细胞单独培养组(D组),每组设2个复孔。共培养120 h后收集各组培养上清液,以备检测IL-17水平。此步骤进行3次,分别使用3次转染实验的DC。
六、IL-23、IL-17 水平检测
步骤四、步骤五收集的细胞培养上清液中IL-23、IL-17水平的检测采用ELISA方法(BioSourceTM,Life Technologies Corporation),操作按试剂盒说明书进行,每一标本设2个复孔。
七、统计学分析
结 果
一、感染后内脏高敏感小鼠模型建立
模型组小鼠感染旋毛虫后8周,肉眼观未见肠道水肿,光学显微镜下未见肠黏膜破坏和明显炎性细胞浸润。结肠气囊扩张压力为40和60 mm Hg(1 mm Hg=0.133 kPa)时,模型组 AWR均分显著高于对照组(40 mm Hg:2.41±0.26 对 1.39±0.16,P<0.01;60 mm Hg:3.23±0.37 对 2.72±0.23,P<0.05),个体最低AWR均分(40 mm Hg:2.28±0.19,60 mm Hg:3.15±0.18)亦显著高于对照组(P<0.05)。 表明肠道感染消退后,小鼠内脏敏感性持续增高,提示造模成功。
二、构建质粒的酶切鉴定
质粒pGenesil-1.1的多克隆位点为:-MluⅠ-hU6 promoter-Insert DNA-SacⅠ-,本研究在插入的目的基因片段里设计有一个SacⅠ酶切位点,且质粒pGenesil-1.1本身就有一个SacⅠ酶切位点,如插入正确,质粒就能被SacⅠ酶切出一条约1000 bp的DNA片段。经SacⅠ酶切鉴定,所构建的小鼠IL-23 shRNA干扰质粒和无关序列shRNA干扰质粒均符合设计要求(见图1)。
三、小鼠IL-23 shRNA干扰质粒的测序分析
测序分析结果显示所构建质粒中小鼠IL-23 shRNA模板DNA序列与设计序列完全一致(见图2)。


四、DC培养上清液IL-23水平
转染小鼠IL-23 shRNA干扰质粒的DC,转染后培养上清液中的IL-23水平较转染前显著降低;转染空脂质体或无关序列shRNA干扰质粒的DC,转染前后IL-23水平无明显变化(见表1)。
表1 DC转染前后培养上清液IL-23水平比较(,pg/mL)
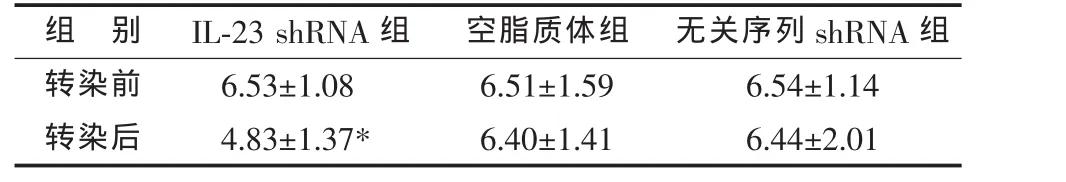
表1 DC转染前后培养上清液IL-23水平比较(,pg/mL)
*与同组转染前比较,P<0.05
五、转染后DC与CD4+T细胞共培养上清液IL-17水平
CD4+T细胞与转染小鼠IL-23 shRNA干扰质粒、空脂质体或无关序列shRNA干扰质粒的DC共培养后,共培养上清液中的IL-17水平均较单独培养的CD4+T细胞显著增高[(12.63±0.63)pg/mL、(14.31±0.44)pg/mL 和(14.22±0.53)pg/mL 对(11.15±0.93)pg/mL,P<0.05],其中IL-23 shRNA组显著低于空脂质体组和无关序列shRNA组(P<0.05),空脂质体组和无关序列shRNA组间差异无统计学意义。
讨 论
PI-IBS是常见功能性肠病IBS的主要亚型,近年研究发现PI-IBS患者的肠黏膜存在由T细胞介导的持续低度炎症[1,2]。DC是肠黏膜免疫系统中最重要的抗原呈递细胞,肠黏膜固有层中存在大量未成熟的DC,摄取抗原后转化为成熟DC。DC在向CD4+T细胞呈递抗原的同时,还能分泌细胞因子诱导CD4+T细胞分化。
CD4+Th细胞作为调节性T细胞中的一个大类,在机体免疫应答、免疫调节等诸多方面发挥重要作用。Th细胞可分为Th1、Th2、Th17三个亚群,三者均由初始型CD4+T细胞分化而成;作为Th17细胞产生的代表性细胞因子,IL-17与自身免疫性疾病密切相关,参与了相关疾病组织炎症的调节[6~8]。研究[9]发现IL-17在T细胞介导的慢性肠道炎症的维持中发挥重要作用。PI-IBS患者的肠黏膜处于持续低度炎症状态,因此Th17细胞可能参与了此种低度炎症状态的维持。
旋毛虫感染后内脏高敏感小鼠模型可模拟人类PI-IBS的自然过程[5],目前已广泛应用于PI-IBS的研究。本课题组的前期研究[4]显示,小鼠感染旋毛虫后2周,肠黏膜可见典型急性炎症改变,结肠气囊扩张AWR评分提示内脏敏感性增高;感染后8周,肠黏膜已无明显炎症改变,但AWR评分提示内脏敏感性持续增高。感染旋毛虫后8周,小鼠肠黏膜固有层DC与脾脏CD4+T细胞共培养能显著促进IL-17分泌,作用强于感染后2周的肠黏膜固有层DC。上述结果表明在急性炎症期,肠黏膜固有层DC能诱导CD4+T细胞分化为Th17细胞并使之活化,随着肠道炎症的自愈,该作用并不减弱,反而进一步增强,即肠道感染消退后,DC仍能持续有效地活化Th17细胞,这一作用可能对PI-IBS肠黏膜免疫系统的持续激活具有重要意义。
关于PI-IBS时肠黏膜固有层DC活化Th17细胞的具体机制,目前仍不甚清楚。研究[10]发现回肠末端菌群可刺激黏膜固有层DC产生IL-23。IL-23是一个由p19亚基和IL-12p40亚基组成的异源二聚体,属于IL-12细胞因子家族成员[11],是诱导初始型CD4+T细胞分化为Th17细胞并分泌IL-17的关键细胞因子[6~8]。推测PI-IBS时肠黏膜固有层DC可能系通过分泌IL-23刺激CD4+T细胞分化,诱导活化Th17细胞,参与肠道感染消退后肠黏膜免疫系统的持续激活。
RNA干扰作为一种高效、特异的基因干预工具,目前已广泛应用于各个研究领域。p19亚基为IL-23发挥特异性作用的关键亚基,外周血DC、极化的Th1细胞和激活的巨噬细胞中均可检测到高水平p19表达。因此本研究以小鼠IL-23基因p19亚基的一段序列为靶点设计、构建了小鼠IL-23 shRNA干扰质粒,以高效、特异地沉默小鼠IL-23基因。酶切鉴定和测序分析显示小鼠IL-23 shRNA干扰质粒构建成功,目的序列完全正确。模拟人类PI-IBS的感染后内脏高敏感小鼠模型肠黏膜固有层DC转染小鼠IL-23 shRNA干扰质粒后,培养上清液中的IL-23水平较转染前显著降低,而转染空脂质体或无关序列shRNA干扰质粒的DC转染前后IL-23水平无明显变化,证明所构建的小鼠IL-23 shRNA干扰质粒能有效抑制肠黏膜固有层DC的IL-23表达。经RNA干扰抑制IL-23表达的DC与CD4+T细胞共培养,虽然亦能促进IL-17分泌,但作用显著弱于IL-23表达未受抑制(转染空脂质体或无关序列shRNA干扰质粒)的DC。由此推测感染后内脏高敏感小鼠肠黏膜固有层DC可能通过分泌IL-23刺激CD4+T细胞分化,诱导活化Th17细胞,参与维持肠道感染消退后肠黏膜免疫系统的持续激活;在PI-IBS肠黏膜免疫系统激活的过程中,可能存在着DC-IL-23-Th17-IL-17这一免疫激活通路,当然这并不是惟一的通路。这一发现对进一步认识PI-IBS的发病机制以及探索新的治疗靶点具有重要意义。
1 Dunlop SP,Jenkins D,Spiller RC.Distinctive clinical,psychological,and histological features of postinfective irritable bowel syndrome[J].Am J Gastroenterol,2003,98(7):1578-1583.
2 Spiller R,Campbell E.Post-infectious irritable bowel syndrome[J].Curr Opin Gastroenterol,2006,22(1):13-17.
3 Dunlop SP,Jenkins D,Spiller RC.Age-related decline in rectal mucosal lymphocytes and mast cells[J].Eur J Gastroenterol Hepatol,2004,16(10):1011-1015.
4 王文峰,龙艳芹,汪欢,等.感染后内脏高敏感小鼠肠道黏膜固有层树突细胞对CD4+T细胞的作用[J].胃肠病学,2010,15(3):147-150.
5 Wheatcroft J, Wakelin D, Smith A, et al.Enterochromaffin cell hyperplasia and decreased serotonin transporter in a mouse model of postinfectious bowel dysfunction[J].Neurogastroenterol Motil,2005,17(6):863-870.
6 Wynn TA.TH-17:a giant step from TH1 and TH2[J].Nat Immunol,2005,6(11):1069-1070.
7 Harrington LE,Hatton RD,Mangan PR,et al.Interleukin 17-producing CD4+effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages[J].Nat Immunol,2005,6(11):1123-1132.
8 Park H,Li Z,Yang XO,et al.A distinct lineage of CD4 T cellsregulatestissueinflammation byproducing interleukin 17[J].Nat Immunol,2005,6(11):1133-1141.
9 Yen D,Cheung J,Scheerens H,et al.IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6[J].J Clin Invest,2006,116(5):1310-1316.
10 Becker C,Wirtz S,Blessing M,et al.Constitutive p40 promoter activation and IL-23 production in the terminal ileum mediated by dendritic cells[J].J Clin Invest,2003,112(5):693-706.
11 Oppmann B,Lesley R,Blom B,et al.Novel p19 protein engagesIL-12p40 to form a cytokine,IL-23,with biological activities similar as well as distinct from IL-12[J].Immunity,2000,13(5):715-725.
猜你喜欢
昆明医科大学学报(2022年3期)2022-04-19
宁夏医学杂志(2020年3期)2021-01-21
散文诗世界(2019年6期)2019-09-10
中国临床医学影像杂志(2019年6期)2019-08-27
意林·全彩Color(2019年7期)2019-08-13
中成药(2018年2期)2018-05-09
中成药(2018年2期)2018-05-09
天然产物研究与开发(2018年4期)2018-05-07
中成药(2017年3期)2017-05-17
西南医科大学学报(2015年1期)2015-08-22
