
GC-MS法测定腌制水产品中3种N-亚硝胺含量
2012-10-21 08:31王秀元蒋玲波张薇英王萍亚
浙江海洋大学学报(自然科学版) 2012年6期
王秀元,赵 华,蒋玲波,方 益,张薇英,王萍亚
(1.浙江海洋学院食品与药学学院、医学院,浙江舟山 316004;2.浙江舟山市质量技术监督检测研究院,浙江舟山 316021)
腌制是一种传统的加工保藏方法,可以提高食品的防腐性能,延长保质期,改善产品风味,其在水产品加工中应用频繁。腌制水产品因其风味独特,深受世界各地人民的青睐[1-3]。但腌制水产品的安全性问题颇具争议,特别是腌制水产品的过程中,会产生含有致癌作用的亚硝酸盐和由此转化生成的N-亚硝胺类化合物[4-6]。相关文献资料表明,人类是N-亚硝胺引起癌症的易感群体[7]。在习惯吃腌制水产品的沿海地方,鼻咽癌和胃癌发病率非常高。针对此现象许多学者研究了某些地区的海产品中硝酸盐、亚硝酸盐、N-亚硝胺的含量调查,结果从腌制水产品中检测到了不同水平的强致癌物质N-亚硝胺[8-10]。
目前国内外对腌制水产品中N-二甲基亚硝胺(NDMA)、N-二乙基亚硝胺(NDEA)的研究较多[11-12],GB2762-2005《食品中污染物限量》规定了腌制生食水产品中NDMA、NDEA的限量标准分别为NDMA≤4 μg/kg,NDEA≤7 μg/kg[13],但常用的方法分析时间长,“气相色谱-热能分析仪”使用率低,因此本研究建立一种适用于腌制水产品中多种N-亚硝胺的高灵敏度快速检测方法。
1 材料与方法
1.1 仪器与试剂
1.1.1 主要仪器
气相色谱-质谱联用仪(GC/MS),Agilent GC7890A/MS5975C,美国Agilent公司;固相萃取装置,美国VISIPRETMDL;高速冷冻离心机,SER JSE09L31,USA;数控超声波清洗器,KQ5200DE,昆山市超声波仪器有限公司。
1.1.2 试剂
标准品:N二甲基亚硝胺(纯度98.0%)由美国CIL提供;N-二乙基亚硝胺(纯度99.9%)、N-二丙基亚硝胺(纯度99.9%)由美国SUPELCO公司提供;二氯甲烷:色谱纯;无水硫酸钠:分析纯。
1.2 试验方法
1.2.1 色谱条件
色谱柱:DB-WAX,30 m×0.250 mm×0.25 μm;进样口温度:250 ℃;载气:He(纯度>99.999%);隔垫吹扫流量:3 mL/min;不分流进样;升温程序:起始温度50℃,保持3 min,以10℃/min升温至110℃,再以15℃/min升温至200℃,再以40℃/min升温至240℃;自动进样,进样量1 μL。
1.2.2 质谱条件
电离方式:电子轰击(EI)离子源;电子能量:70 eV;电子倍增器电压:1 671 V;离子源温度:230℃;四极杆温度:150℃;接口温度:240℃;SIM采集模式,扫描质量范围:m/z 40~350;溶剂延迟:4 min;采样频率:2。
上述条件下,3种N-亚硝胺的保留时间和定性定量离子见表1。

表1 3种N-亚硝胺物质的保留时间和定性定量离子表Tab.1 Retention time and GC/MS acquisition parameters for three N-nitrosamines
1.2.3 标准溶液制备
分别准确称取3种N-亚硝胺标准品0.1 g(精确到0.000 1 g),用二氯甲烷定容至50 mL,得到N-亚硝胺标准储备液,浓度为2.0 mg/mL。准确移取上述标准储备液,以二氯甲烷逐级稀释配制N-亚硝胺:10 ng/mL、50 ng/mL、100 ng/mL、200 ng/mL、1 000 ng/mL 系列混合标准溶液,按 1.2.1 色谱条件和 1.2.2 质谱条件上机进样,进样量1 mL,以混合标准品浓度为横坐标,对应的峰面积为纵坐标,绘制标准曲线。
1.2.4 样品溶液制备及前处理
称取粉碎的腌制水产品样品20.0 g,置于50 mL具塞离心管中,加入二氯甲烷30 mL,于IKA振药器上涡旋1 min,保持温度在30℃以下超声提取30 min,低温条件下离心10 min,分出有机相溶剂,再提取2次,每次约20 mL二氯甲烷,合并有机溶剂相,通过固相萃取技术净化萃取样品,经无水硫酸钠脱水,于40℃水浴下真空浓缩至近干,定容1.0 mL,备用。
2 结果与讨论
2.1 色谱柱的优化
考察了不同极性的气相色谱柱(非极性柱DB-5MS:30 m×0.250 mm×0.25 μm、强极性柱DB-WAX:30 m×0.250 mm×0.25 μm)对待测N-亚硝胺类物质的分离效果。结果表明:选用非极性柱时,色谱峰分离度也不好,N-二甲基亚硝胺的色谱峰与溶剂峰分不开;而选用强极性柱DB-WAX时,获得的色谱峰峰形对称、定量准确、重现性好、分离效果好。因此,本试验选用DB-WAX作为色谱柱。
2.2 柱温升温速率的优化
由于溶剂峰对对待测物质出峰的影响,因此需要优化柱温的升温速率,以获得最佳分离效果。选择柱温起始温度为50℃,分别比较了升温速率为15℃/min、10℃/min、5℃/min时分离效果,实验结果显示:当升温速率为15℃/min时,溶剂峰与N-二甲基亚硝胺的色谱峰无法明确分出;当升温速率为10℃/min时,能够有效分离,而继续降低升温速率为5℃/min时,色谱峰的分离效果没有明显改善,因此选择升温速率为10℃/min。当柱温升高到110℃时,N-二甲基亚硝胺的色谱峰已出峰,因此为了提高分析速度,将升温速率提高为15℃/min。因此,最终选择柱温为:起始温度50℃,保持3 min,以10℃/min升温至110℃,之后以15℃/min升温至200℃,再以40℃/min升温至240℃。
按照上述优化条件,选择电子轰击电离源(EI),电子能量为70 eV,采集模式为SIM,对3种N-亚硝胺混合标样分析,得到标准品的保留时间和特征离子见表1,气相色谱-质谱总离子流图如图1所示(浓度为200 ng/mL)。
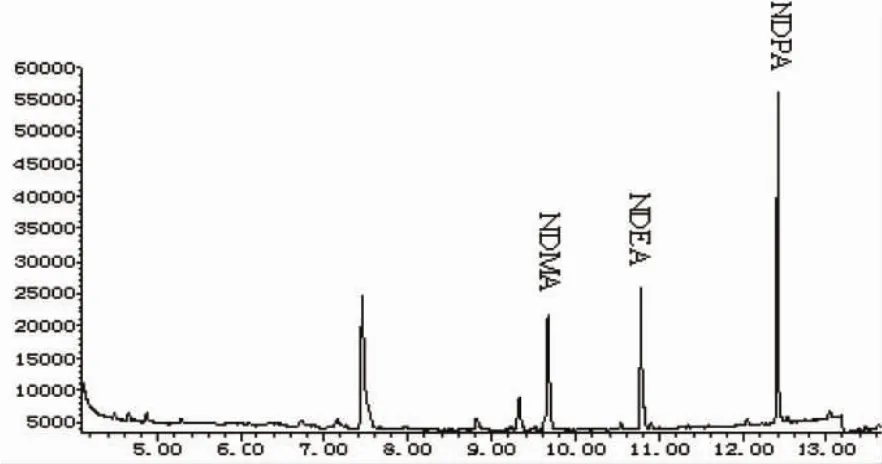
图1 3种N-亚硝胺标准品的气相色谱-质谱总离子流图(浓度为200 ng/mL)Fig.1 Total ion current chromatogram of three N-nitrosamines(c:200 ng/mL)
2.3 前处理条件的选择
2.3.1 样品提取方法的优化
考察了分别选用水蒸气蒸馏法和超声振荡提取法对样品进行前处理,结果表明:超声振荡提取法获取N-亚硝胺比水蒸气蒸馏法提取更完全,且操作简捷,因此,选择超声振荡法提取样品中的N-亚硝胺类物质。
2.3.2 固相萃取小柱的优化
本实验比较了5种固相萃取小柱净化的效果。样品加入适量N-亚硝胺混合标样溶液,方法同1.2.4。通过比较每个萃取小柱所萃取的3种亚硝胺的回收率来确定最佳的萃取小柱,平行3次试验,结果取平均值,作表2。由表2可以看出,选取活性炭固相萃取小柱效果较好,因此,本试验采用活性炭萃取柱。
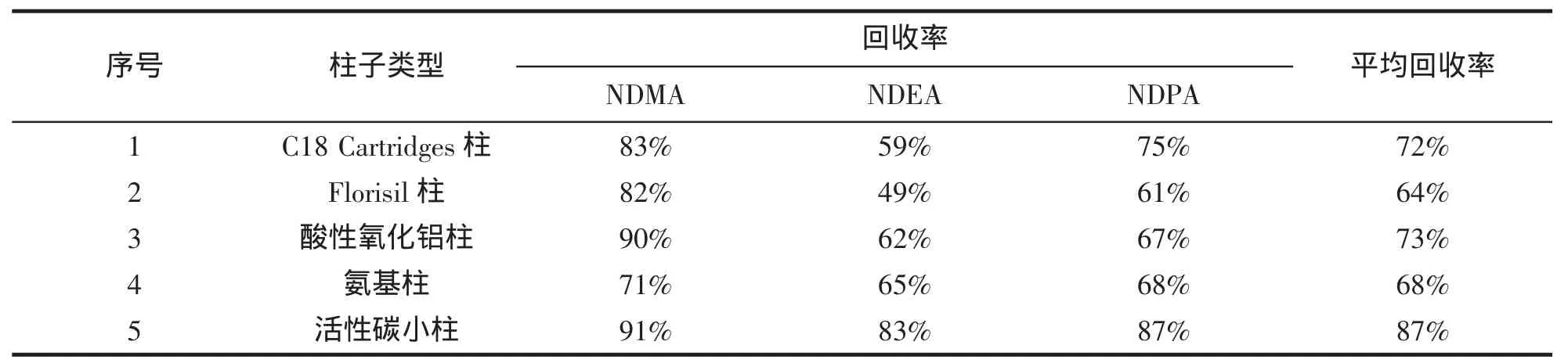
表2 5种固相萃取小柱萃取效果比较图Tab.2 Comparison of clean-up effects of five SPE cartridges
2.4 工作曲线及检测限
本方法采用外标法定量,配制5种N-亚硝胺的质量浓度为10 ng/mL、50 ng/mL、100 ng/mL、200 ng/mL、1 000 ng/mL系列混合标准溶液,利用定量离子峰面积绘制标准曲线。每个质量浓度的样品测定3次,以目标物的峰面积Y对其质量浓度X进行回归分析,得到各N-亚硝胺的线性方程、相关系数和线性范围见表2,3种化合物的选择性离子色谱图和扫描质谱图如图2~4所示。
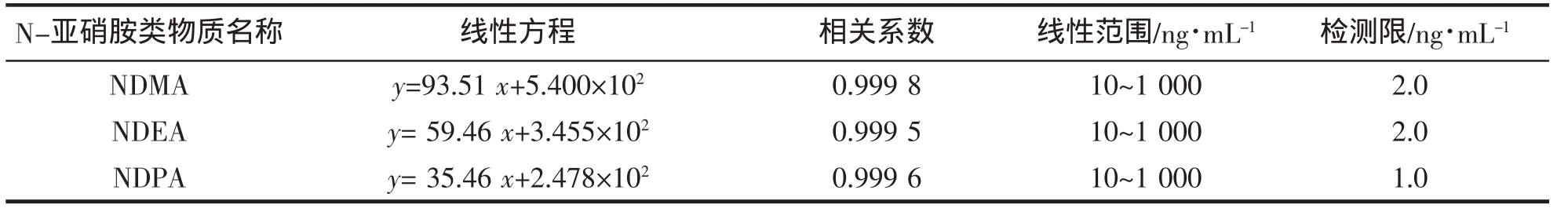
表3 3种挥发性N-亚硝胺线性方程、相关系数、线性范围及检测限Tab.3 Regression equationgs,correlation coefficients,linear ranges and LOQs for three N-nitrosamines
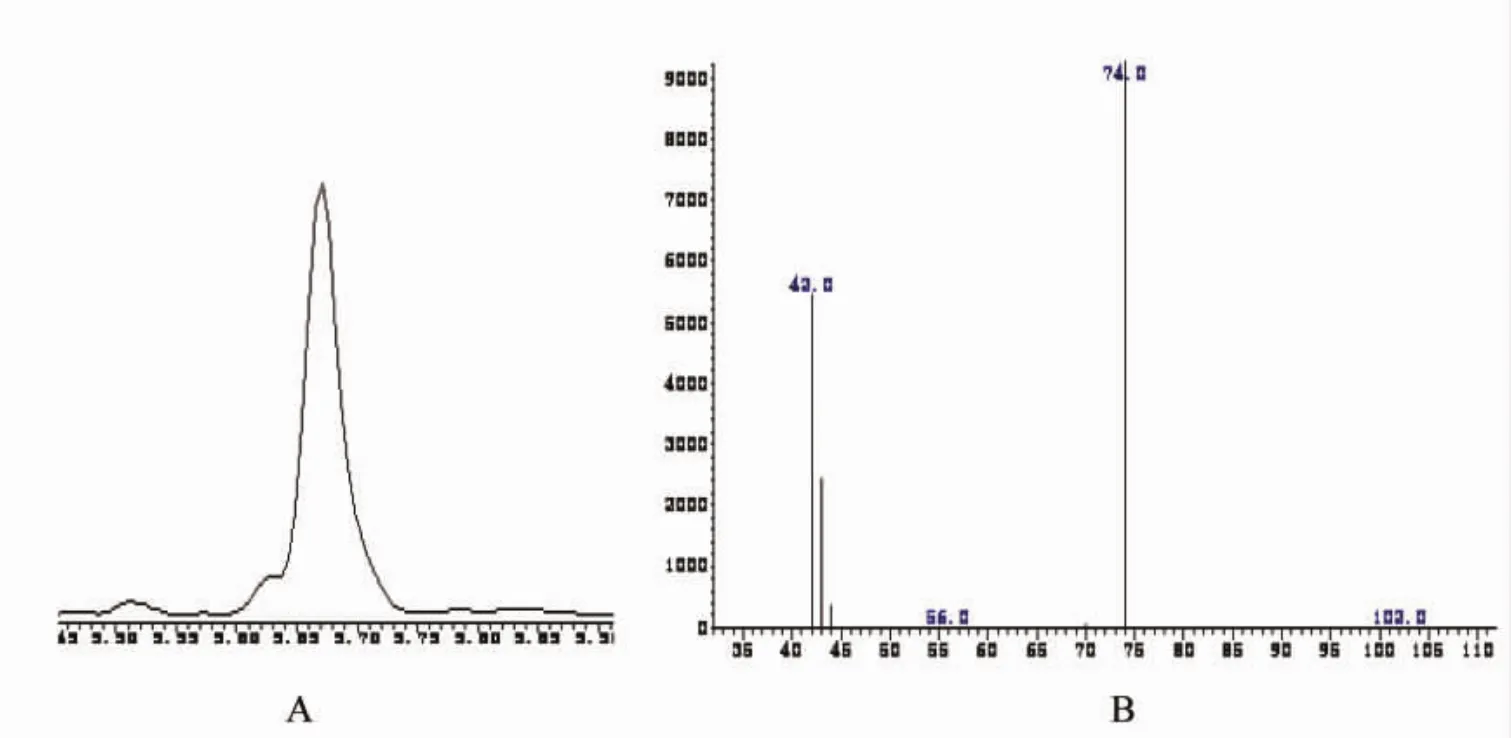
图2 NDMA选择性离子色谱图A和扫描质谱图B(9.671 min)Fig.2 NDMA chromatogram(A)and mass spectrogram(B)of MIB(9.671 min)

图3 NDBA选择性离子色谱图A和扫描质谱图B(10.775 min)Fig.3 NDBA chromatogram(A)and mass spectrogram(B)of MIB(10.775 min)
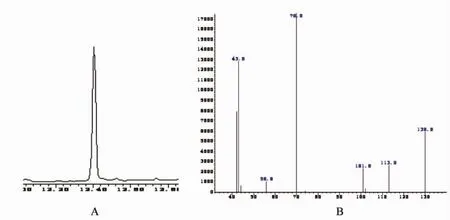
图4 NDPA选择性离子色谱图A和扫描质谱图B(12.405 min)Fig.4 NDPA chromatogram(A)and mass spectrogram(B)of MIB(12.405 min)
由表3可知,本方法重现性好,线性范围广,相关系数可达0.999 5以上,检测灵敏度高,检测限:NDPA为1 ng/mL,NDMA、NDEA为2 ng/mL,达到ppb级。
2.5 回收率及精密度试验
选择腌制泥螺样品进行加标回收试验,加混合标样200 ng/mL,各进行6次样品测定,同时做空白样试验,检测其本底成分,结果见表4。

表4 回收率及精密度(n=6)Tab.4 Precision and recovery for three N-nitrosamines(n=6)
由表4可知,本试验采用的样品处理方法,能够达到国家标准规定的要求。
2.6 样品测定分析
取3种比较常见的舟山腌制水产品样品:鳕鱼、鮟鱇鱼、泥螺,分别编号:①、②、③,按1.2项试验方法分别测定挥发性N-亚硝胺的含量,每个样品平行3次,结果见表5,其中腌制泥螺的选择离子色谱图如图5。
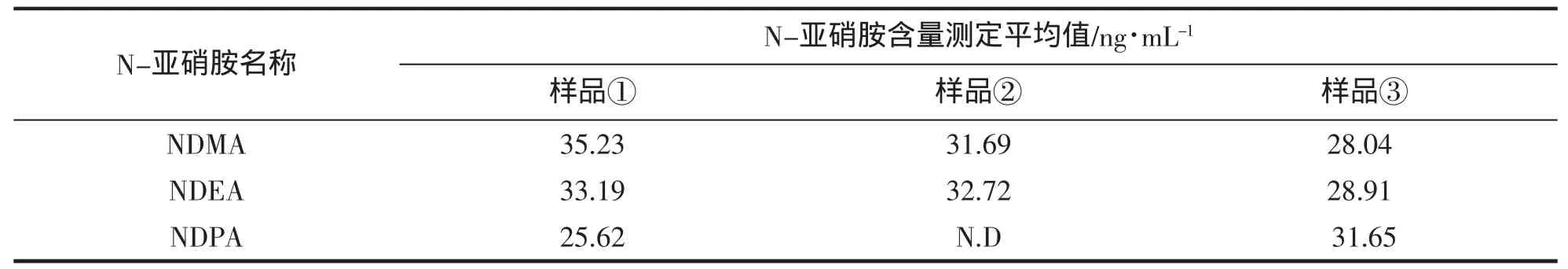
表5 实际样品中3种挥发性N-亚硝胺含量测定Tab.5 Determination results of three N-nitrosamines in real samples
3 结论
本研究建立了一种适用于气相色谱-质谱联用法测定腌制水产品中3种N-亚硝胺的分析方法。比较了不同提取方法、不同固相萃取小柱、不同色谱柱对分离检测的影响,结果表明:以二氯甲烷作为溶剂提取腌制水产品中的N-二甲基亚硝胺(NDMA)、N-二乙基亚硝胺(NDEA)、N-二丙基亚硝胺(NDPA)3种化合物,采用超声振荡法提取,选用活性炭萃取小柱,DB-WAX(30 m×0.250 mm×0.25 μm)色谱柱,分离效果良好。该方法前处理更简单快速,且易于操作,重现性好,回收率可观,在86%~99%之间,线性范围10~1 000 ng/mL,方法灵敏度高,检测限:NDPA为1 ng/mL,NDMA、NDEA为2 ng/mL,达到ppb级,并在实际样品分析中应用良好,可适用于腌制水产品中N-亚硝胺物质含量的测定分析。

图5 腌制泥螺的选择离子色谱图Fig.5 SIM chromatogram of salted snail
[1]HONIKEL K.The use and control of nitrate and nitrite for the processing of meat products[J].Meat Science,2007,78:68-76.
[2]陈维娟.咸鱼深加工工艺探讨[J].中国水产,2002(4):74-75.
[3]李基银.腌菜质量与卫生[M].北京:轻工业出版社,1988:128-172.
[4]YURCHENKO S,MOLDER U.The occurrence of volatile N-nitrosaminesin Estonian meat products[J].Food Chemistry,2007,100:1 713-1 721.
[5]杨 华,马俪珍,王 瑞,等.肉制品中N-亚硝胺及亚硝酸盐测定及其相关性分析[J].保鲜与加工,2006,35(4):21-23.
[6]王绍云.腌鱼、腌肉中亚硝酸盐及硝酸盐含量的测定[J].贵州师范大学学报:自然科学版,2004,22(2):87-89.
[7]吴永宁.现代食品安全科学[M].北京:化学工业出版社,2003:248-259.
[8]马俪珍,南庆贤,方长法.N-亚硝胺类化合物与食品安全性[J].农产品加工学刊,2005(12):8-14.
[9]方长发,马俪珍,刘会军,等.固相微萃取技术及其在N-亚硝胺分析中的应用[J].肉类研究,2008(4):49-53.
[10]樊丽琴,杨贤庆,陈胜军,等.腌制水产品中N-亚硝基化合物的研究进展[J].食品工业科技,2009(4):360-363.
[11]魏法山,徐幸莲,周光宏.挥发性N-亚硝基化合物的分析方法[J].食品科学,2008,29(7):484.
[12]吉林省卫生防疫站.GB/T 5009.26-2003食品中N-亚硝胺类的测定[S].北京:中国标准出版社,2004.
[13]中国疾病预防控制中心营养与食品安全所,卫生部卫生监督中心.GB 2762-2005食品中污染物限量[S].北京:中国标准出版社,2006.
猜你喜欢
少年文艺(2022年8期)2022-07-08
化学工程师(2022年3期)2022-04-19
食品与发酵工业(2020年16期)2020-09-03
中成药(2017年10期)2017-11-16
中国经济周刊(2017年6期)2017-03-21
家庭百事通·健康一点通(2016年12期)2016-12-29
科技与创新(2015年20期)2015-10-29
科技与企业(2015年20期)2015-10-21
中国医疗美容(2015年1期)2015-07-12
科技视界(2013年24期)2013-11-12
