
表皮生长因子受体突变细胞系H1650耐药机制探讨
2012-09-10 06:59韩瑞丽王小丽钟殿胜赵娟陈哲孙琳琳王竞张金棒
中国肺癌杂志 2012年12期
韩瑞丽 王小丽 钟殿胜 赵娟 陈哲 孙琳琳 王竞 张金棒
肺癌主要分为非小细胞肺癌(non-small cell lung cancer, NSCLC)和小细胞肺癌(small cell lung cancer,SCLC),其中80%-85%为NSCLC。尽管现代医学的诊疗技术已经有了相当的进展,采取了手术为主、放化疗等综合治疗手法,肺癌总的5年生存率仍只有13%-15%[1]。研究[2,3]发现,表皮生长因子受体(epidermal growth factor receptor, EGFR)在50%-90%的NSCLC患者中高表达,参与肿瘤的血管新生、迁移和粘附过程,其扩增和突变已被认为是肺部肿瘤发生的主要机制之一。厄洛替尼(Erlotinib, Tarceva)是EGFR酪氨酸激酶抑制剂(tyrosine kinase inhibitor, TKI),通过特异性结合在EGFR胞内酪氨酸激酶区域抑制EGFR活化而发挥抗瘤作用[4]。本研究探讨了NSCLC细胞系EGFR基因的表达水平及其抑制剂Erlotinib对NSCLC细胞系的毒性作用。
1 材料与方法
1.1 材料 人NSCLC细胞系A549、H460、H157、H1299、H1792、CALU-1、H1650、H1975和HCC827购自美国模式培养物集存库(American Type Culture Collection,ATCC)。RPMI-1640培养基、新生小牛血清购于GIBCO公司;Trizol Reagent购自美国Invitrogen公司;反转录试剂、SYBR Premix Ex Taq购自TAKARA公司,MTT试剂盒购于美国Promega公司,p-AKT、p-ERK、AKT、ERK、PTEN抗体均购于美国Cell Signaling Technology公司,GAPDH抗体购于美国Santa Cruz公司,二抗HRP-羊抗兔IgG、二抗羊抗鼠IgG购于北京中杉生物技术有限公司,PVDF膜购于美国Amersham Biosciences。
1.2 方法
1.2.1 细胞培养 用含10%小牛血清的RPMI-1640培养基、100 U/mL青霉素、100 U/mL链霉素配制成1640完全培养基,37oC、5%CO2、饱和湿度的培养箱中传代培养,0.125%胰酶消化传代,3 d-4 d传一代。所有实验均采用对数生长期细胞。
1.2.2 Real-time RT-PCR 用Trizol试剂提取处于对数生长期细胞总RNA,反转录mRNA为cDNA,使用realtime RT-PCR技术检测基因的表达情况。PCR引物序列为:EGFR:Forward:GCGTTCGGCACGGTGTATAA;Reverse:GGCTTTCGGAGATGTTGCTTC;参照基因GAPDH:Forward:GGAGTCAACGGATTTGGTCG;Reverse:CTGATTGGAGGGATCTCG,扩增长度为240 bp。PCR扩增体系20 μL,第一步:预变性 95oC、30 s、1个循环,第二步:PCR反应:95oC、5 s,60oC、34 s,40个循环。以GAPDH为内参照,2-ΔΔCT法计算EGFR基因表达差异。
1.2.3 MTT法检测细胞存活率 取对数生长期细胞,常规胰酶消化制成单细胞悬浮液,以每孔约5×103个细胞接种于96孔板,每孔200 μL,培养过夜后弃去原液,分别加入终浓度为1×10-3μM/L、1×10-2μM/L、1×10-1μM/L、1 μM/L、10 μM/L和20 μM/L的Erlotinib,对照组加入等量的培养基,每组设4个复孔,72 h后进行MTT试验,并绘制细胞生长曲线。
1.2.4 Western blot方法 按照文献[5],提取细胞总蛋白,BCA法测定蛋白浓度,取50 μg蛋白上样,十二烷基硫酸钠-聚丙烯酰胺凝胶电泳,转膜、封闭,一抗4oC过夜,二抗室温1 h,使用ECL化学发光试剂工作液进行蛋白信号检测,GAPDH作为蛋白加样对照。
2 结果
2.1 EGFR野生型NSCLC细胞系中EGFR mRNA表达水平及与Erlotinib细胞毒性相关性的研究
2.1.1 EGFR野生型NSCLC细胞系中EGFR mRNA表达水平利用real-time RT-PCR技术检测了6种EGFR野生型NSCLC细胞系中EGFR mRNA表达水平。实验结果显示(图1),H157表达水平最低;A549和CALU-1表达水平相对较低;H460和H1792表达水平较高;H1299表达水平最高,约为H157的2,621倍,为EGFR高表达的细胞系。

图 1 EGFR野生型NSCLC细胞系中EGFR mRNA表达水平Fig 1 The epidermal growth factor receptor (EGFR) mRNA expression level in EGFR wild-type non-small cell lung cancer (NSCLC) cells

图 2 MTT方法检测EGFR野生型NSCLC细胞对Erlotinib药物敏感性Fig 2 The measurement of cytotoxicity for EGFR wild-type NSCLC cells to Erlotinib by MTT assay

图 3 Western blot方法检测EGFR的突变情况Fig 3 The EGFR mutations in H1975, H1650 and HCC827 cells

图 4 MTT检测EGFR突变细胞系对Erlotinib药物敏感性Fig 4 The measurement of cytotoxicity for EGFR-mutant NSCLC cells to Erlotinib by MTT assay
2.1.2 Erlotinib对上述细胞系的毒性作用 6株细胞系分别在7个浓度梯度的Erlotinib培养液中培养72 h,利用MT方法检测细胞毒性,结果显示(图2):随着药物浓度的倍增,Erlotinib对各细胞系的生长抑制作用并没有明显增强,均表现为明显的耐药性。其中在1 μM Erlotinib的浓度,H157细胞系的生长抑制率约为10%,A549和Calu-1的生长抑制率约为0,H460和H1792细胞系的生长抑制率分别约为12%和15%,而高表达EGFR的H1299细胞系的生长抑制率为0;在10 μM Erlotinib的浓度,H157细胞系的生长抑制率为24%,A549和Calu-1细胞系的生长抑制率分别约为10%和2%,H460和H1792的生长抑制率分别约为20%和21%,H1299的生长抑制率8%。上述结果表明,EGFR野生型NSCLC细胞系对Erlotinib耐药,且Erlotinib的细胞毒性与EGFR mRNA表达水平高低无关。
2.2 Erlotinib对EGFR突变型NSCLC细胞系毒性作用的研究 肺癌中EGFR突变多见于外显子18-21,即胞内酪氨酸激酶编码区,最常见突变形式包括外显子19的E746-A750del和外显子21的L858R点突变,这两种突变约占EGFR突变的85%-90%[6],发生这两种突变的肿瘤细胞对EGFR-TKIs敏感,称为活化突变。部分肿瘤细胞可发生二次突变,最常见二次突变为外显子20的T790M突变,为耐药突变[7]。
H1650和HCC827细胞系均为EGFR外显子19的E746-A750del突变,H1975细胞系为外显子21 L858R点突变,但同时伴有外显子20的T790M二次突变。我们利用Western blot验证了上述3种NSCLC细胞系中EGFR的突变情况(图3)。
3株细胞系分别在7个浓度梯度Erlotinib的培养液中培养72 h,细胞毒性实验结果显示(图4),Erlotinib能够明显抑制HCC827的生长,呈明显的浓度相关性,IC50为0.03 μM;H1975对Erlotinib高度耐药,这些结果均与预期的一致。但实验结果显示,H1650细胞系对Erlotinib相对耐药,约为HCC827的167倍,与预期的推测不一致。
2.3 Erlotinib对H1650细胞信号传导途径的影响 EGFR主要的信号传导通路包括:Ras/Raf/MEK/ERK通路和PI3K/AKT通路。EGFR与EGFR-TKI结合后,抑制EGFR本身磷酸化,从而抑制其下游相应的信号蛋白的磷酸化。
我们用10 μM Erlotinib处理HCC827和H1650细胞2 h,结果显示,在HCC827细胞中,AKT和ERK的磷酸化水平均明显下调(图5),提示Erlotinib可以明显抑制其细胞内EGFR下游的信号通路;在H1650细胞中,p-ERK水平呈明显下降,但AKT的磷酸化水平无明显下降(图5)。

图 5 Western blot方法检测Erlotinib处理HCC827和H1650后p-ERK、p-AKT的表达水平Fig 5 HCC827 and H1650 cells were treated with Erlotinib for 2 h,Anti-p-AKT and anti-p-ERK antibody was used to detect AKT and ERK phosphorylation, with GAPDH as the loading control.
Sos等[8]发现在H1650细胞中有PTEN的缺失。我们实验结果也证实了在H1650细胞中存在PTEN的缺失(图6)。
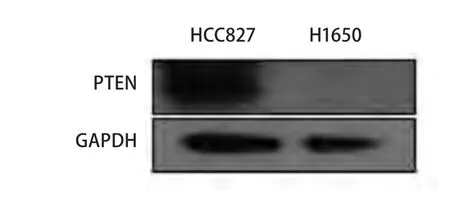
图 6 Western bot检测HCC827和H1650细胞中PTEN的表达水平Fig 6 No PTEN protein detected in H1650 cells
蛋白酪氨酸磷酸酶(phosphatase and tensin homolog deleted on chromosometen, PTEN)是具有蛋白与脂质磷酸酯酶活性的双特异性磷酸酯酶,能特异地使磷脂酰肌醇-3,4,5-三磷酸3'位脱磷酸,抑制Akt的磷酸化[9]。PTEN的缺失可以导致Akt的活化,从而可以解释Erlotinib为什么不能抑制H1650细胞AKT磷酸化水平,但可抑制ERK的磷酸化。为了进一步探讨H1650耐药与PTEN表达缺失的相关性,我们用10 μM LY294002(PI3K抑制剂)处理H1650及HCC827细胞2 h,进而检测p-AKT的水平,结果显示(图7)HCC827细胞的p-AKT明显抑制,但对H1650细胞的p-AKT抑制作用不明显,进一步证实了因为PTEN的缺失,H1650的AKT磷酸化水平不受其上游调节;因为LY294002对Ras/Raf/MEK/ERK通路无作用,所以两种细胞系的p-ERK水平未被抑制(图7)。

图 7 Western blot检测经LY294002处理后的HCC827和H1650细胞系的p-AKT、p-ERK表达水平Fig 7 HCC827 and H1650 cells were treated with LY294002 (PI3K inhibitor) for 2 h, Anti-p-AKT and anti-p-ERK antibody was used to detect AKT and ERK phosphorylation, with GAPDH as the loading control.
综合上述结果,H1650细胞系对Erlotinib相对耐药可能与PTEN缺失导致AKT信号传导通路异常活化有关,而与Ras/Raf/MEK/ERK信号途径无关。
3 讨论
肺癌是死亡率最高的癌症之一,其中约80%-85%为NSCLC[10],由于约70%的患者在就诊时已处于晚期,失去了手术的机会,且NSCLC对铂类等化疗药物的反应差,其5年生存率只有15%左右[11]。
EGFR是原癌基因C-erbB1的表达产物,属于酪氨酸激酶生长因子受体家族成员之一,EGFR主要的信号传导通路包括Ras/Raf/MEK/ERK通路和PI3K/AKT通路。在肿瘤细胞中,EGFR基因突变和扩增可使EGFR酪氨酸激酶不恰当激活,促进肿瘤的血管生成和肿瘤细胞的增殖、粘附、侵袭和转移。研究[12]显示,NSCLC伴转移的患者中,60%以上存在EGFR过度表达,且与这些患者的预后密切相关,因此以EGFR为靶点的抗癌治疗日益受到关注。EGFR突变主要发生在胞内酪氨酸激酶编码区,多发生于基因外显子18、19、20和21,与EGFRTKIs敏感性相关的主要是位于外显子18、21的点突变和外显子19的缺失突变。其中,外显子21的点突变,使EGFR蛋白中该位点的氨基酸由亮氨酸转变为精氨酸(L858R);外显子19第746-750位密码子的缺失(19 exon E746-A750del)导致EGFR蛋白中氨基酸序列丢失,改变了受体络氨酸激酶ATP结合槽的角度。
EGFR-TKIs通过与ATP竞争,结合于EGFR-TK胞内端的催化区域(EGFR结构域中高度保守的ATP结合位点),阻止EGFR的自磷酸化及下游的信号传导。EGFRTKIs代表药物,Gefitinib或Erlotinib,作为二线药物已应用于标准化疗无效的NSCLC;对于存在EGFR突变的晚期NSCLC患者,EGFR-TKI一线化疗药物应用于临床已经得到了专家的共识[13]。但随着EGFR-TKI临床应用的日益增多,其耐药现象已成为临床工作中一大难题。 引起耐药的原因多种多样,主要分为原发性和继发性耐药[14],原发性耐药机制包括KRAS突变[15];继发性耐药机制主要包括EGFR外显子20的T790M突变(苏氨酸转变为甲硫氨酸)和C-MET扩增两种,约占所有耐药机制的70%,尚有30%-40%患者耐药机制不清楚[16]。
本研究显示,EGFR野生型NSCLC细胞系对Erlotinib均耐药,且Erlotinib的药物敏感性与EGFR的mRNA表达水平无关,但与EGFR突变类型相关;具有EGFR外显子19缺失突变的H1650细胞对Erlotinib相对耐药。Guo等[17]发现在H1650细胞株中,PTEN蛋白表达缺失,因此对AKT的抑制作用消失。Yamamoto等[18]用Gefitinib处理PC-9细胞系(存在EGFR外显子19缺失突变)7个月后诱导出耐药细胞系,经验证发现,耐药细胞系是由于PTEN蛋白表达缺失导致对Gefitinib耐药的;他们用免疫组化的方法对4例Gefitinib获得性耐药的肺癌患者进行了治疗前后PTEN蛋白表达水平的比较,其中3例患者PTEN表达水平明显低于治疗前,考虑PTEN蛋白表达水平的缺失与Gefitinib获得性耐药有关。我们的实验结果也验证了在H1650细胞中存在PTEN蛋白表达的缺失,同时发现Erlotinib虽然可以抑制H1650细胞中的p-ERK水平,但p-AKT的水平无明显变化。因此推测,H1650对Erlotinib耐药可能与PTEN缺失有关。进一步应用PI3K抑制剂LY294002处理H1650细胞,发现其也不能抑制H1650细胞的p-AKT水平,进一步证实H1650细胞p-AKT水平不受其上游调节,其对Erlotinib相对耐药与PTEN表达缺失导致AKT信号传导通路异常活化有关,而与Ras/Raf/MEK/ERK信号途径无关。
此外,亦有文献报道H1650对Erlotinib的耐药可能与抑制BIM上调[19]和肿瘤细胞从上皮细胞向间充质细胞转化(epithelial mesenchymal transition, EMT)[20]有关,这些均有待于我们在未来的研究中进一步证实。
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
现代农村科技(2022年1期)2022-01-21
中国循证心血管医学杂志(2021年10期)2021-11-05
食品界(2021年7期)2021-07-19
中国生殖健康(2020年4期)2021-01-18
中国生殖健康(2018年4期)2018-11-06
杂草学报(2017年4期)2017-04-12
医学研究杂志(2015年9期)2015-07-01
癌变·畸变·突变(2015年4期)2015-02-27
湖北农业科学(2014年11期)2014-09-10
