
氧化损伤在HIV相关神经认知损害中的作用
2012-09-05 10:23张玉林乔录新宋凤丽徐树莹陈德喜
首都医科大学学报 2012年5期
丁 渭 张玉林 乔录新 宋凤丽 徐树莹 陈德喜
(北京市肝病研究所,首都医科大学附属北京佑安医院,北京100069)
对感染人类免疫缺陷病毒(human immunodeficiency virus,HIV-1)的患者的脑组织进行尸检可以发现在中枢神经系统存在神经细胞和非神经细胞的凋亡,尤其在HIV相关性痴呆。尽管神经相关痴呆的发病频率非常高,而最严重的神经认知障碍由于抗反转录病毒试剂的应用已开始下降,但在艾滋病患者中仍然存在大量的神经认知合并症,这些都归类为HIV相关的神经认知障碍。
HIV-1感染的非神经细胞,如巨噬细胞和小胶质细胞,对转录反式激活因子(trans-activator of transcription,Tat),病毒蛋白 R(viral protein regulatory,Vpr)和HIV-1包膜糖蛋白gp120通过免疫刺激产生神经毒素诱导的神经毒性是必需的[1-3]。这些神经毒素是兴奋性毒素,与谷氨酸受体结合,主要是C-X-C趋化因子受体4(C-X-C chemokine receptor 4,CXCR4)和 CC趋化因子受体5(C-C chemokine receptor 5,CCR5),使这些受体过度激活。这些过度激活的受体能够激活细胞内钙离子信号通路并使细胞内活性氧增加[3-6]。线粒体活性氧生成后Tat蛋白诱导细胞质游离钙离子水平逐步升高[5-6]。因此,HIV-1蛋白,尤其是gp120,Tat和Vpr能够刺激活性氧(reactive oxygen species,ROS)的产生,表明在艾滋病患者中凋亡样神经元死亡是由活性氧介导的。迄今为止发现的活性氧诱导的所有病变中,DNA和RNA病变最丰富的是8-氧鸟嘌呤(8-hydroxyguanine,8-oxoG)。为了检测额叶皮质中DNA氧化损伤在感染了HIV-1的中枢神经系统神经元损伤中是否起重要作用,本研究在感染了HIV的中枢神经系统对氧化核DNA损伤和线粒体DNA损伤进行了研究。此外,我们试图在艾滋病患者尸检的额叶组织中检测8-氧鸟嘌呤的水平,以此作为艾滋病毒导致的神经系统疾病包括HIV相关的神经认知功能障碍患者活性氧水平的生物标记。
1 材料与方法
1.1 抗体、主要设备和试剂
本研究中用到的抗体主要有:抗-8-氧鸟嘌呤(8-oxoG)单克隆抗体(Trevigen公司,美国)、抗-8-氧-20脱氧鸟苷糖甘酶1(OGG1)多克隆抗体(Novus Biologicals,Littleton,CO,美国)、FITC标记的抗兔 IgG和Cy3标记的抗鼠IgG(Sigma公司,美国)。其他主要试剂有:二氢乙锭(Dihydroethidium)(Invitrogen公司,美国)。病毒载量通过 Nuclisens EasyQ分析仪(Nuclisens EasyQ HIV-1 1.1,biomerieux bv)检测,而CD4细胞计数通过流式细胞仪检测(BD FACSCalibur,美国)。
1.2 HIV相关的神经认知障碍评估
HIV相关神经认知功能障碍采用国际获得性免疫缺陷综合征(acquired immunodeficiency syndrome,AIDS)痴呆量表进行评分(IHDS),包括记忆-记录-回忆3个步骤,主要测试运动速度和心理运动速度[7]。IHDS总分12分。IHDS评分低于或等于10分考虑为HIV-1相关神经认知紊乱(HIV-1 associated neurocognitive disorders,HAND),得分越低越严重。
1.3 尸检脑组织标本
本研究中的10例额叶脑组织来源于河南省1994和1995年因商业性献血感染了HIV-1的一组艾滋病死亡患者。10例尸检脑组织标本中,5例生前依据IHDS考虑为HAND,另外5例为非HAND。5例正常额叶皮质脑组织标本来源于首都医科大学附属北京朝阳医院神经外科脑部肿瘤患者的癌旁脑组织碎片。研究经过首都医科大学附属北京佑安医院伦理委员会审核批准。组织采用10%的甲醛固定。
1.4 免疫荧光染色
组织玻片保存于-80℃。1×triton穿孔15 min后,1×PBS洗涤15 min×2次;1%BSA+2%羊血清+1×PBS 37℃封闭1 h;1∶1 000浓度抗-8-oxoG,抗OGG1抗体(封闭液稀释)4℃过夜;1×PBS洗涤15 min×3次;1∶1 000浓度对应的Cy3或FITC标记IgG二抗(封闭液稀释)37℃温育1 h;1×PBS洗涤20 min×3次;DAPI封片后于德国Leica-dm500b型正向荧光显微镜下阅片并用Xillix数码相机拍摄相关图像(每个点至少选取3个视野,取平均数)。8-oxoG的阳性水平可通过8-oxoG阳性细胞核/DAPI阳性细胞核比率算得,OGG1相关密度可用Image Pro Plus 6.0软件进行分析。
1.5 DNA提取和实时定量PCR
使用中国天根公司DNA提取试剂盒,按照使用说明提取基因组DNA。通过实时定量PCR相对定量的方法,我们分析了这些尸检组织中线粒体DNA的拷贝数变化,包括5例HAND、5例非HAND和5例正常对照。首先,我们对实际DNA含量与qPCR实验所获得的DNA拷贝数之间关系进行验证,建立标准曲线(图1A)。实时定量PCR检测采用已经成熟的探针法[8],相对定量。采用 Taqman7900HT系统进行实时定量PCR扩增线粒体DNA。所有的引物探针均由Invitrogen公司合成(上海,中国),所用序列详见表1。每个标本作3个复孔,并设立阴性和阳性对照。引物和探针在反应中的终浓度为250和300 nm。反应程序依次为:95℃ 3 min,95℃15 s 40个循环,60℃ 1 min。利用Microsoft Excel软件对数据进行分析处理。
1.6 线粒体DNA D-loop区的克隆测序
我们通过PCR的方法从前面提取的脑组织标本基因组DNA中扩增线粒体DNA D-loop区,再通过BioEdit软件将克隆测序所得标本D-loop区序列与Gen-Bank线粒体DNA标准序列(NC_012920)进行核苷酸进化距离分析。根据标准的方法学,使用高保真的Taq多聚酶(Invitrogen公司,美国),用10 ng总DNA进行PCR扩增。PCR产物根据厂商说明书连接到pGEM-18T载体(Tankra公司,中国)。所有的序列由中国Biotake公司通过ABI3130序列仪进行测序。使用NCBI的BLAST和BioEdit软件进行序列分析。使用7.0.5.3版本BioEdit软件的Clustal W多序列比对程序进行多序列的比对。
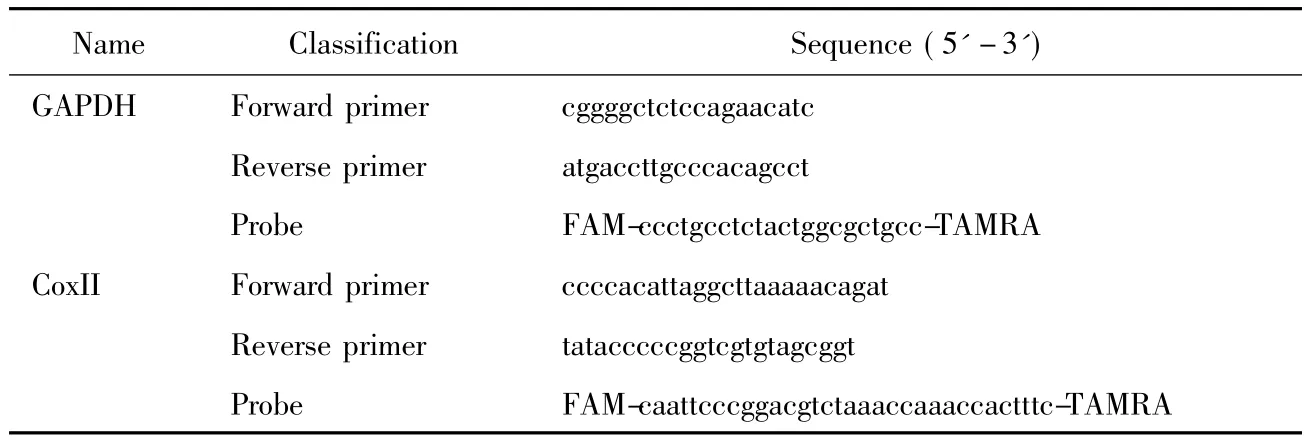
表1 实验所用引物和探针序列Tab.1 Primer and probe sequences used in the experiment
1.7 通过K562细胞检测AIDS患者脑脊液中超氧化物
基于荧光技术二氢乙啶(HE)常被用于检测细胞和组织中的超氧化物。HE在细胞质中本身会发出蓝色荧光(吸收/发射波长:355/420纳米),一旦被氧化形成2-羟基乙啶,与DNA嵌合后会发出红色荧光(吸收/发射波长:518/605纳米)[9]。只有当它脱氢(氧化)成2-OH-E+后才可以与DNA嵌合。为了进一步验证AIDS患者中枢神经系统氧化损伤情况,本研究中,利用稳定表达EGFP绿色荧光的人类白血病K562-EGFP细胞系和艾滋病患者(包括HAND和非HAND患者)新鲜脑脊液共同孵育15 min。然后利用流式细胞技术检测K562-EGFP细胞HE的水平。人类白血病K562-EGFP细胞一直由本实验室保存,用含有10%胎牛血清,100U/mL青霉素和100 μg/mL链霉素的RPMI1640培养基在加湿的5%CO2培养箱中,接种于6孔板中培养。1×105K562-EGFP细胞与200 μL的艾滋病患者脑脊液共同孵育15 min为了检测AIDS患者脑脊液中超氧化物。K562-EGFP细胞在0.03%H2O2中孵育5 min作为阳性对照,同时将没有做任何处理的K562-EGFP细胞作为阴性对照。去除培养基,加入1×PBS稀释的储存浓度为10 mg/mL的HE[于二甲基亚砜(dimethyl sulfoxide,DMSO)中储存](Polysciences公司,美国),使终浓度为 50 μg/mL。避光,37 ℃孵育10 min。最后用BD FACSDiva流式细胞仪对细胞进行分析(BD FACS CantoTMⅡ)。HE在490 nm波长处被激发,在620 nm波长处可被检测到。储存10 000个细胞的数据,然后用CellQuest软件进行分析。
1.8 数据分析
每个样品均分析了8-oxoG和OGG1,在额叶皮质组织中随机选取了显微镜视野(20×)的10个细胞进行计数分析。所有统计分析均使用SPSS18.0统计软件,采用单因素方差分析进行比较,以P<0.05为差异有统计学意义。
2 结果
2.1 患者一般情况和临床资料
10名艾滋病患者,这些患者分别在2006年和2007年去世。其中有7名男性和3名女性,平均年龄为32.9岁(8~56岁)。所有的患者终末期CD4计数都很低,平均为31.4(13~56)×106/L。这些患者的HIV-1亚型为HIV-1B亚型。最后一次记录的平均血浆病毒载量是102 400(28 000-219 000)拷贝/mL;脑脊液(cerebrospinal fluid,CSF)病毒载量未知。所有患者未见中枢神经系统机会性感染,但是有3例患者合并肺结核,1例患者合并肺囊虫感染,1例患者合并淋巴瘤,还有1例患者眼底巨细胞病毒感染,4例患者没有任何机会性感染。IHDS测试表明5例患者有神经认知障碍(≤10分)。5例对照患者没有任何机会性感染、HIV-1感染、未进行过化疗和放射治疗。
2.2 AIDS患者大脑皮质线粒体DNA检测
HAND组和非HAND组线粒体DNA拷贝数下降到正常对照组的(0.69±0.18)倍和(0.74±0.15)倍(图1B)。这表明,HIV-1慢性感染者的脑组织中线粒体DNA含量明显下降。但是,在非HAND组和HAND组之间脑组织中线粒体DNA含量差异无统计学意义。
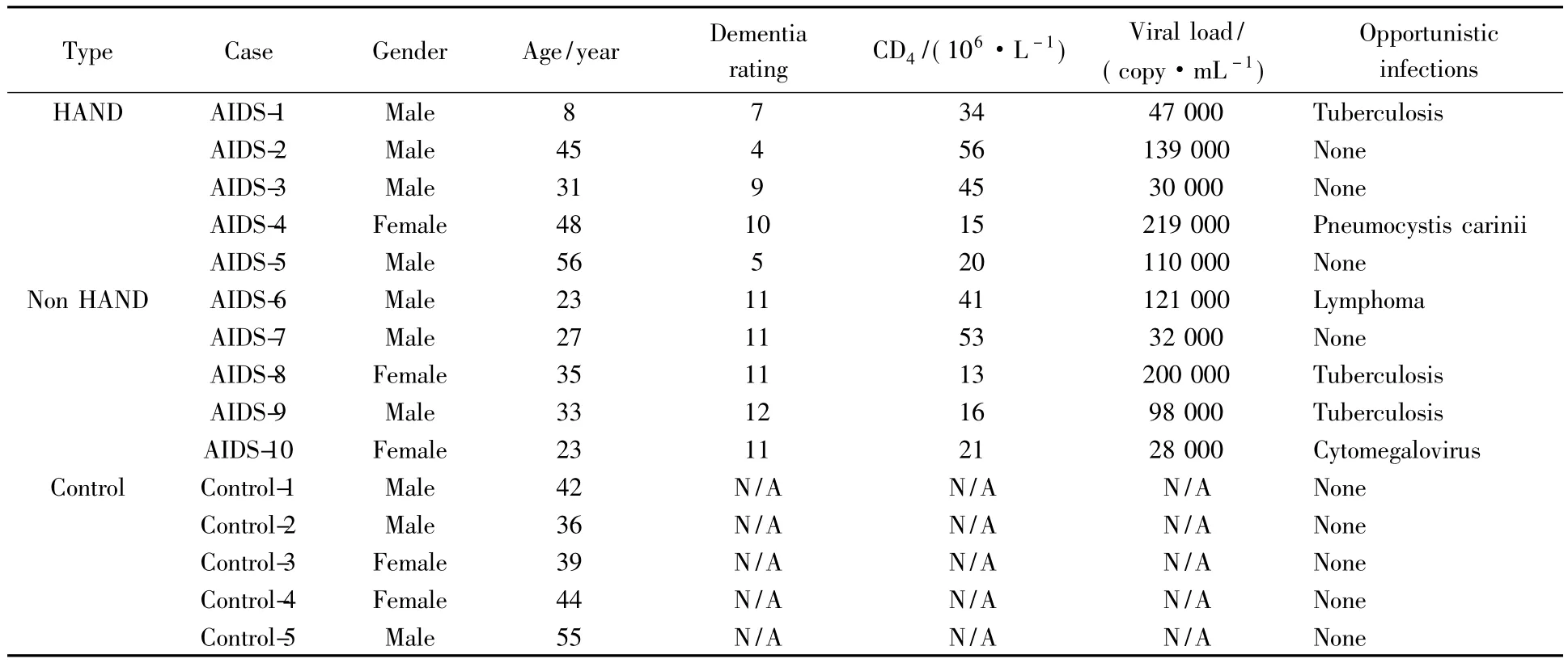
表2 患者一般情况和临床资料Tab.2 General information:dementia rating CD+4T cell,viral load and opportunistic infections of the 10 AIDS patients and 5 control cases

图1 HIV-1感染者大脑皮质mtDNA缺失Fig.1 Depletion of mtDNA was detected in frontal cortex autopsy tissue from HIV-1 infected patients
2.3 DNA氧化损伤标志物8-oxoG在AIDS患者尸检皮质脑组织中显著增加
HAND组和非HAND组患者脑组织中8-氧鸟嘌呤的表达水平均增加,而且HAND组和非HAND组脑组织8-氧鸟嘌呤的表达水平增加更加明显。比较而言,对照组额叶皮质组织中几乎检测不到8-氧鸟嘌呤的表达(图2A)。对3次独立实验结果进行统计分析,结果显示HAND组有近45%8-oxoG阳性细胞,非HAND组有近30%8-oxoG阳性细胞,而对照组只有4%8-oxoG阳性细胞(图2B)。
发现AIDS患者尸检组织中OGG1水平明显低于对照组,但HAND和非HAND患者之间OGG1水平差异无统计学意义(图2C,D)。
2.4 艾滋病患者尸检组织额叶皮质中线粒体DNA突变增加
AIDS患者尸检组织中D-loop区DNA突变水平明显增加。核苷酸进化距离是0.047 1(0.012 2-0.122 7);而对照组的5份标本中只有2人存在缺失(D310C),没有突变(图3A)。进一步分析显示,与对照组相比,HAND尸检组织在线粒体DNA D-loop区突变率明显增高(图3B),单核苷酸突变数量在HAND组织中是39,非HAND组织中是13,而在对照组标本中仅有2个。HAND患者额叶皮质高水平的单核苷酸突变可能与线粒体功能障碍相关,进而增加了活性氧的水平。
2.5 艾滋病患者脑脊液中超氧化物增加
能够代表脑脊液中超氧阴离子水平的HE的检出率随着HAND的进展而逐渐增加:阴性对照为4%,非HAND组为28% ~40%,HAND组为37% ~65%,阳性对照0.3%H2O2组为99%(图4)。
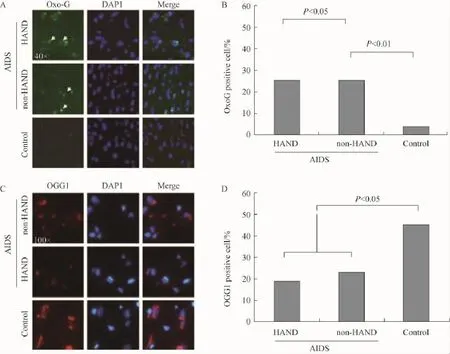
图2 HIV-1感染患者大脑皮质脑细胞核DNA 8-oxoG氧化损伤和OGG1蛋白低表达Fig.2 High levels of 8-oxoG oxidative DNA damage in nuclear DNA and low levels of OGG1 protein co-exist in frontal cortex autopsy tissue from HIV-1 patients

图3 大脑皮质脑组织mtDNA D-loop非编码区突变分析.Fig.3 Mutation assay of the noncoding region of the mtDNA D-loop in frontal cortex autopsy issue from HIV-1 infected samples and controls
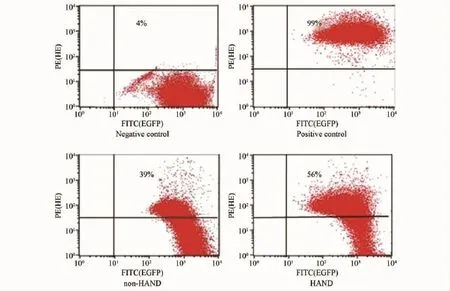
图4 AIDS患者脑脊液超氧阴离子水平Fig.4 Level of superoxide in cerebrospinal fluid from AIDS Patients
3 讨论
HIV-1感染患者的尸检脑组织可见神经细胞和非神经细胞的凋亡,尤其在HIV相关痴呆症患者[10-12]。早期研究[13-16]表明 HIV-1 对神经突触的损害和诱导慢性细胞凋亡可导致大脑局部(如海马和基底节区)神经元的死亡,继而引起认知和运动功能障碍。艾滋病患者脑组织中神经凋亡的机制非常复杂,目前主流观点认为由于分裂细胞(单核巨噬细胞和巨噬细胞)感染了HIV-1后释放神经毒性物质,继而产生神经毒性[17]。因此,HIV-1蛋白 gp120、Tat和Vpr能够刺激活性氧的产生这一现象表明艾滋病患者神经元凋亡或死亡可能是由活性氧介导的。在细胞内,酶促和非酶促抗氧化反应组成一个复杂的防御系统以对抗各种氧化应激。然而,HIV-1蛋白在诱导过量活性氧产生的同时,还抑制机体抗氧化系统功能,从而进一步加重氧化损伤对神经元的毒性作用。
众所周知,线粒体DNA氧化损伤和线粒体DNA缺失是非常严重的损伤,能够导致线粒体呼吸功能下降和细胞凋亡。此外,慢性神经元凋亡是HAND主要的病理改变。本研究中,HIV-1感染时间均较长。随着HIV相关的神经认知功能障碍的进展,活性氧的产生和氧化线粒体DNA的损伤(突变和缺失)能够干扰线粒体氧化磷酸化,从而进一步诱导神经细胞凋亡,加速HIV相关神经认知功能障碍的进展[18-22]。迄今为止发现的活性氧诱导的所有病变中,DNA和RNA病变最丰富的是8-氧鸟嘌呤,而且可能作为活性氧水平有效的生物标志。既往研究[23]表明氧化应激可能在HIV感染的神经病变中发挥重要作用,HIV-1感染患者的CD+4T细胞中8-氧鸟嘌呤水平增加,DNA糖基化酶活性减低;而CD+8T细胞中8-氧鸟嘌呤的水平与健康对照组相似。我们对HIV-1感染患者尸检组织额叶皮质的研究发现8-氧鸟嘌呤染色阳性的细胞占所有DAPI阳性细胞的45%,但是在健康对照组中只有4%。HIV-1感染的患者尸检脑组织中氧化损伤DNA的积累以及AIDS患者中枢神经系统高水平的活性氧可能在神经退行性疾病中发挥关键作用。本研究的结果表明活性氧和OGG1能影响艾滋病患者中枢神经系统中8-氧鸟嘌呤的聚集。另外的试验[24]证明8-oxo-G和OGG1表达主要在神经元细胞。OGG1可启动针对DNA损伤的碱基切除修复。对老龄化和神经退行性疾病的研究[18]发现OGG1活性与8-氧鸟嘌呤积聚呈负相关。
临床研究[25]发现,阿尔茨海默病等神经退行性疾病以及老龄化可见线粒体DNA突变率和缺失率增加。HIV-1感染和抗病毒治疗所用的核苷类似物反转录抑制剂是线粒体DNA突变和线粒体功能障碍的两个重要原因[26-27]。心肌细胞中特异表达HIV Tat能够破坏其线粒体;此外,HIV Tat的过表达能够抑制线粒体超氧化物歧化酶的表达[28]。在HIV-1感染早期,病毒即能进入中枢神经系统。此后在HIV-1感染大脑的慢性病程中,神经免疫的激活以及病毒蛋白的直接毒性能够增加神经元和胶质细胞氧化应激的水平,导致大量的活性氧产生,包括羟自由基、超氧阴离子和单态氧等[29]。正如我们初步结果所揭示的那样,脑脊液中的超氧化物随着HAND的发展而增加,而线粒体基质中高水平的活性氧使得细胞线粒体DNA突变率大大增加。此外,HIV-1感染本身就可以产生氧化应激,并且刺激线粒体谷氨酰胺酶过表达,从而在HAND病程中诱导谷氨酸介导的神经细胞凋亡通路[10,29-30]。因此,我们对线粒体DNA突变和缺失是否与HAND相关很感兴趣,最终研究结果显示HIV-1感染的患者尸检脑组织中存在高水平的线粒体DNA突变和缺失。有研究[31]显示HIV-1感染和HIV-1蛋白Tat、Vpr可诱导线粒体产生活性氧,从而影响HAND的进程。我们的研究结果显示HAND和非HAND患者线粒体DNA缺失显著不同,表明HIV-1相关中枢神经系统病理过程是非常复杂的。众所周知,HIV-1感染的巨噬细胞和胶质细胞释放的促炎性细胞因子,HIV-1蛋白和兴奋性氨基酸能够直接或间接刺激神经元产生活性氧,从而损伤细胞大分子物质,导致脂质过氧化、蛋白质氧化和DNA氧化损伤。在HAND患者脑脊液中检测到的高水平的活性氧表明,中枢神经系统活性氧与HAND的发展相关。氧化应激的慢性积累会诱导神经元凋亡,导致神经退行性疾病的发生和发展。因此,确定氧化应激在诱导核DNA氧化损伤和线粒体DNA突变,从而导致神经细胞凋亡中的作用非常重要。同时,这些结果表明,抗氧化剂治疗不乏为预防和治疗HAND的一种有益的选择。
[1]Buzy J,Brenneman D E,Pert C B,et al.Potent gp120-like neurotoxic activity in the cerebrospinal fluid of HIV-infected individuals is blocked by peptide T[J].Brain Res,1992,598(1-2):10-18.
[2]Jones G J,Barsby N L,Cohen E A,et al.HIV-1 Vpr causes neuronal apoptosis and in vivo neurodegeneration[J].J Neurosci,2007,27(14):3703-3711.
[3]Yeung M C,Pulliam L,Lau A S.The HIV envelope protein gp120 is toxic to human brain-cell cultures through the induction of interleukin-6 and tumor necrosis factor-alpha[J].AIDS,1995,9(2):137-143.
[4]Kaul M,Lipton S A.Chemokines and activated macrophages in HIV gp120-induced neuronal apoptosis[J].Proc Natl Acad Sci USA,1999,96(14):8212-8216.
[5]Norman J P,Perry S W,Reynolds H M,et al.HIV-1 Tat activates neuronal ryanodine receptors with rapid induction of the unfolded protein response and mitochondrial hyperpolarization[J].PLoS One,2008,3(11):e3731.
[6]Perl A,Banki K.Genetic and metabolic control of the mitochondrial transmembrane potential and reactive oxygen intermediate production in HIV disease[J].Antioxid Redox Signal,2000,2(3):551-573.
[7]Sacktor N C,Wong M,Nakasujja N,et al.The International HIV dementia scale:a new rapid screening test for HIV dementia[J].AIDS,2005,19(13):1367-1374.
[8]Wu Y,Li N,Zhang T,et al.Mitochondrial DNA base excision repair and mitochondrial DNA mutation in human hepatic HuH-7 cells exposed to stavudine[J].Mutat Res,2009,664(1-2):28-38.
[9]Kaufman J,Jing J.China and AIDS-the time to act is now[J].Science,2002,296(5577):2339-2340.
[10]Allard J P,Aghdassi E,Chau J,et al.Effects of vitamin E and C supplementation on oxidative stress and viral load in HIV-infected subjects[J].AIDS,1998,12(13):1653-1659.
[11]Garden G A,Morrison R S.The multiple roles of p53 in the pathogenesis of HIV associated dementia[J].Biochem Biophys Res Commun,2005,331(3):799-809.
[12]Petito C K,Roberts B.Evidence of apoptotic cell death in HIV encephalitis[J].Am J Pathol 1995,146(5):1121-1130.
[13]Kaul M.HIV's double strike at the brain:neuronal toxicity and compromised neurogenesis[J].Front Biosci,2008,13:2484-2494.
[14]Bassiri A,Holden J,Wong M.A case of fulminant human immunodeficiency virus dementia[J].Clin Infect Dis,1995,21(5):1313-1314.
[15]Chiodi F.Tissue lesions in AIDS patients involve both the lymphoid and the nervous system[J].AIDS,1995,9 Suppl A:S41-48.
[16]Kaul M,Lipton S A.Mechanisms of neuronal injury and death in HIV-1 associated dementia[J].Curr HIV Res,2006,4(3):307-318.
[17]Ozdener H.Molecular mechanisms of HIV-1 associated neurodegeneration[J].J Bio Sci,2005,30(3):391-405.
[18]Bolin C M,Basha R,Cox D,et al.Exposure to lead and the developmental origin of oxidative DNA damage in the aging brain[J].FASEB J,2006,20(6):788-790.
[19]Eugenin E A,King J E,Nath A,et al.HIV-tat induces formation of an LRP-PSD-95-NMDAR-nNOS complex that promotes apoptosis in neurons and astrocytes[J].Proc Natl Acad Sci USA,2007,104(9):3438-3443.
[20]Lipton S A.Similarity of neuronal cell injury and death in AIDS dementia and focal cerebral ischemia:potential treatment with NMDA open-channel blockers and nitric oxide-related species[J].Brain Pathol,1996,6(4):507-517.
[21]O'donnell L A,Agrawal A,Jordan-Sciutto K L,et al.Human immunodeficiency virus(HIV)-induced neurotoxicity:roles for the NMDA receptor subtypes[J].J Neurosci,2006,26(3):981-990.
[22]Saitoh A,Fenton T,Alvero C,et al.Impact of nucleoside reverse transcriptase inhibitors on mitochondria in human immunodeficiency virus type 1-infected children receiving highly active antiretroviral therapy[J].Antimicrob Agents Chemother,2007,51(12):4236-4242.
[23]Aukrust P,Luna L,Ueland T,et al.Impaired base excision repair and accumulation of oxidative base lesions inT cells of HIV-infected patients[J].Blood,2005,105(12):4730-4735.
[24]Boiteux S,Radicella J P.Base excision repair of 8-hydroxyguanine protects DNA from endogenous oxidative stress[J].Biochimie,1999,81(1-2):59-67.
[25]Reeve A K,Krishnan K J,Turnbull D M.Age related mitochondrial degenerative disorders in humans[J].Biotechnol J.2008,3(6):750-756.
[26]Casula M,Bosboom-Dobbelaer I,Smolders K,et al.Infection with HIV-1 induces a decrease in mtDNA[J].J Infect Dis,2005,191(9):1468-1471.
[27]Kohler J J,Lewis W.A brief overview of mechanisms of mitochondrial toxicity from NRTIs[J].Environ Mol Mutagen,2007,48(3-4):166-172.
[28]Raidel S M,Haase C,Jansen N R,et al.Targeted myocardial transgenic expression of HIV Tat causes cardiomyopathy and mitochondrial damage[J].Am J Physiol Heart Circ Physiol,2002,282(5):H1672-1678.
[29]Pocernich C B,Sultana R,Mohmmad-Abdul H,et al.HIV-dementia,Tat-induced oxidative stress,and antioxidant therapeutic considerations[J].Brain Res Brain Res Rev,2005,50(1):14-26.
[30]Tian C,Erdmann N,Zhao J,et al.HIV-infected macrophages mediate neuronal apoptosis through mitochondrial glutaminase[J].J Neurochem,2008,105(3):994-1005.
[31]Opii W O,Sultana R,Abdul H M,et al.Oxidative stress and toxicity induced by the nucleoside reverse transcriptase inhibitor(NRTI)-2',3'-dideoxycytidine(ddC):relevance to HIV-dementia[J].Exp Neurol,2007,204(1):29-38.
猜你喜欢
遗传(2022年6期)2022-06-21
影像研究与医学应用(2020年17期)2020-08-20
国际呼吸杂志(2019年20期)2019-11-23
化工生产与技术(2016年5期)2016-11-07
读者(2016年18期)2016-08-23
中国运动医学杂志(2016年3期)2016-07-10
山东医药(2015年38期)2015-12-07
食品工业科技(2014年13期)2014-03-11
中国医学科学院学报(2013年3期)2013-03-11
食品科学(2013年24期)2013-03-11
