
硅(100)表面组装膜的第一性原理研究①
2012-08-21 01:28:10史立秋息明东马清祥
佳木斯大学学报(自然科学版) 2012年3期
史立秋, 息明东, 张 琳, 马清祥
(1.佳木斯大学,黑龙江佳木斯154007;2.佳木斯龙江福浆纸有限公司,黑龙江佳木斯154000)
利用半导体材料硅片为基底自组装有机功能膜的技术是一项很有前景的微纳结构制备技术[1-3].由于这类自组装膜(SAMs)的有序性和常温下的高稳定性,尤其是所制得的功能膜具有纳米尺度结构和界面性质,故很受研究者的青睐.因此,从微观尺度上研究硅表面组装膜前后的分子构型和能量变化进而了解SAMs稳定存在的状态显得尤为重要.
近年来随着理论和数值算法的飞速发展,使得基于密度泛函理论的第一性原理方法成为凝聚态物理、量子化学和材料科学中的常规计算研究手段[4-6].计算所得的结果不仅能用于解释实验结果,而且还有可能可靠地预言材料的很多性质,并在某些情况下导致实验方面的重要发现[7].单晶硅表面有机膜的理论研究已有报道[8-10],但是关于硅表面自组装芳香烃重氮盐膜的计算研究尚未见报道,本章从理论角度,采用第一性原理计算对基于机械-化学法在硅(100)表面制备的芳香烃膜(C6H4NO2)的组装行为进行微观尺度的研究.
1 模拟计算
1.1 计算方法
本文计算根据量子化学密度泛函理论(DFT),使用 Accelery公司的 Material Studio软件中CASTEP(Cambridge Sequential Total Energy Package)计算模块进行理论计算,泛函采用广义梯度近似(GGA),局部泛函采用PBE(Perdew-Burke-Ernzerhof functional),平面波截止能 Ecut设为300eV.
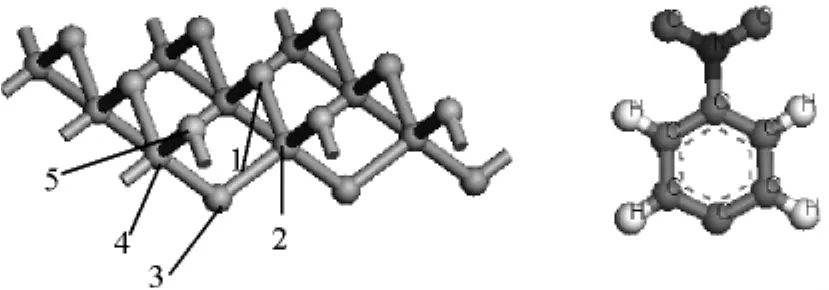
图1 单晶硅(100)晶面及C6 H4 NO2活性分子侧视图
1.2 模型建立
Si(100)晶面计算模型建立条件:沿单晶硅(100)方向切割,深度3层,超晶胞取原始晶胞,真空层厚度15Å.在Si(100)晶面所选取的超晶胞模型中,包含27个Si原子,表面包含9个Si原子.
芳香烃重氮盐计算模型的建立条件:由于计算软件的限制,建立模型时直接建立成断开氮氮三键后的一端为硝基的苯环(C6H4NO2)即可.
计算C6H4NO2与Si(100)晶面相互作用的模型选取:在结构优化完成的Si(100)晶面上选取中间硅原子,把优化好的分子根置于其上方.图1是Si(100)晶面、C6H4NO2模型的侧视图.
2 结果与讨论
2.1 结构优化
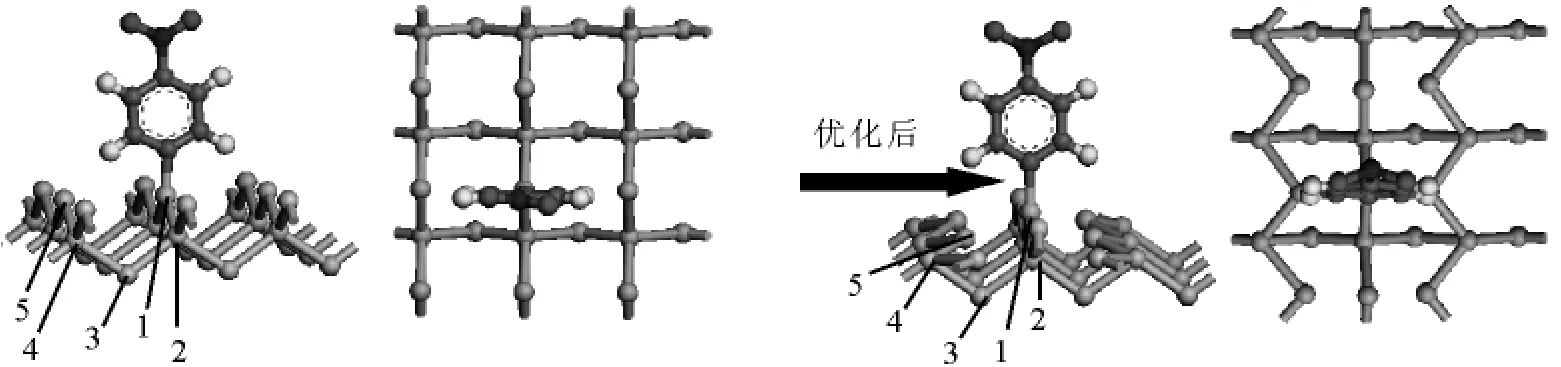
图2 Si(100)晶面结合C6H4NO2活性分子结构
图2是 Si(100)晶面结合一个活性分子C6H4NO2优化前后的侧视图和俯视图.表1分别是Si(100)晶面优化前、优化后、结合一个活性分子C6H4NO2后部分硅原子的键角和键长数据.按结构理论推断单晶硅中每个Si以sp3杂化轨道与另外四个Si原子形成四面体的空间结构,Si—Si键长 d=2.352Å,Si—Si—Si键角为 109.471°.当晶体沿某一方向切割后得到不同表面,晶体表面的原子结构明显地不同于体相的原子结构,其原因是固体中每个原子都贡献出相同数目的电子和周围其它原子形成化学键,从而使固体中原子结合在一起.但是固体表面上的原子由于其一面的原子的化学键被切断,因而具有多余的未配对的价电子形成悬挂键,导致表面的结构与晶体体相内部产生差异.从表1可知结构优化后的键长和键角数据都发生了变化,其数据都与理论值不同.
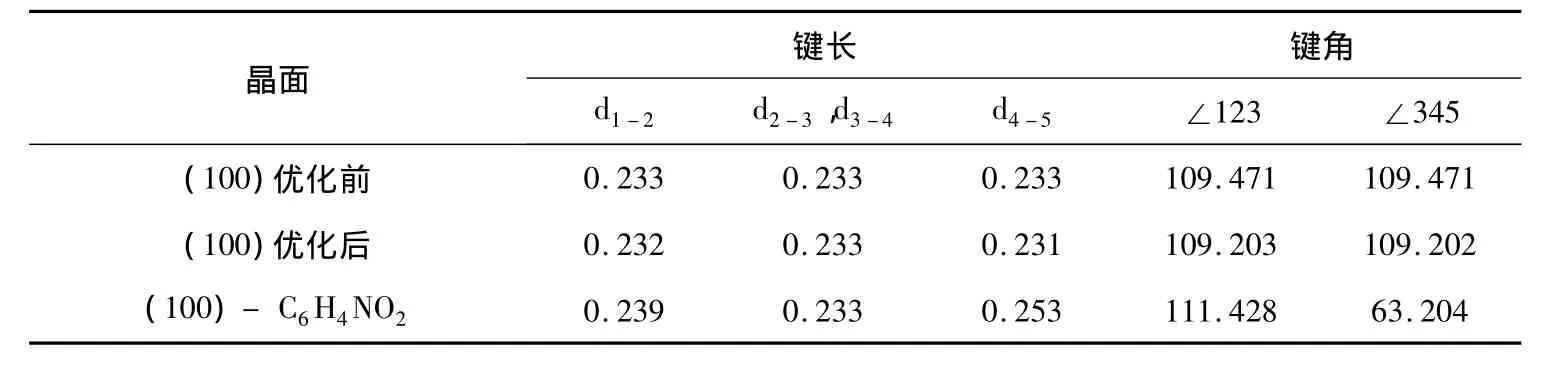
表1 Si(100)晶面优化前后和结合一个C6 H4 NO2前后部分键长(nm)和键角数值(°)
2.2 结合能计算
表2是Si(100)晶面结合一个C6H4NO2前后的总能量,Si原子上结合一个C6H4NO2后能量变低,是由于形成了新的化学键的原因.能量降低的大小可用结合能来表示.当结合能数值为负数时表示释放出能量,体系的稳定性升高.结合能数值越小反应前后能量降低得越多,越有利于反应的发生.结合一个C6H4NO2后,体系总能量降低,降低值就是形成Si—C化学键释放出的能量即结合能为-101.95eV,表明(100)晶面可以组装上芳香烃重氮盐膜,组装后体系的稳定性升高.

表2 Si(100)晶面结合一个C6 H4 NO2分子前后的总能量和结合能
3 结论
本文采用第一性原理方法计算了单晶硅(100)面与芳香烃重氮盐分子组装前后的键长、键角和能量变化.组装前后Si原子间的键长、键角发生了变化,这主要是由于引入带硝基的苯环所致,这些变化的大小从能量的角度看都是影响自组装反应能否进行的因素.结合一个C6H4NO2后体系总能量降低,降低值就是形成Si—C化学键释放出的能量即结合能为-101.95eV,表明硅(100)表面容易组装上芳香烃重氮盐膜,组装后体系的稳定性升高,膜的稳定性较好.
[1]Wagner P.,Nock S.,Spudich J.A.,et al..J.Struc.Bio[J].1997,119:189 -201.
[2]Dusastre V..Nature[J].2000,406:31 -35.
[3]Brent A.W.,Michael J.M.,Ian A.M.,et al..Appl.Phys.Lett[J].2003,82:808 - 810.
[4]Lopinski G.P.,Wayner D.D.M.,Wolkow R.A..Nature[J].2000,406:48 -51.
[5]Sieval A.B.,Opitz R.,Maas H.P.A.,et al..Langmuir[J].2000,16:5688-5695.
[6]Zhang L.,Carman A.J.,Casey S.M..J.Phys.Chem[J].2003,107:8424-8432.
[7]Wei Z.G.,Zhang H.X.,Li Q.S.,et al.Chem JChinese U[J].2008,29:824 -826.
[8]Travis L.N.,Jiang G.L.,Yit Y.L.,et al.Langmuir[J].2001,17:5889-5900.
[9]Yit Y.L.,Travis L.N.,Reija M.,et al.Langmuir[J].2002,18:4840-4846.
[10]Yit Y.L.,Jonathan J.F.,Li Y.,et al.J.Am.Chem.Soc[J].2005,21:2093 -2097.
猜你喜欢
中学化学(2024年3期)2024-06-30 15:19:19
大学物理(2022年9期)2022-09-28 01:10:52
高中数理化(2022年16期)2022-09-14 13:57:06
吉林大学学报(理学版)(2021年3期)2021-05-26 02:24:00
物理通报(2020年7期)2020-07-01 09:28:02
青岛大学学报(工程技术版)(2019年2期)2019-09-10 07:22:44
中学生数理化(高中版.高考理化)(2019年6期)2019-06-22 09:55:38
中国循环杂志(2015年10期)2015-12-24 03:29:56
原子与分子物理学报(2015年3期)2015-11-24 12:49:34
文理导航(2015年26期)2015-09-29 14:12:24
