
液液萃取-GC-MS法测定水中硝基苯质量控制指标研究*
2012-07-02 01:30:02胡冠九李娟袁力夏新
化学分析计量 2012年2期
胡冠九,李娟,袁力 夏新
(国家环境保护地表水环境有机污染物监测分析重点实验室,江苏省环境监测中心,南京 210036) (中国环境监测总站,北京 100012)
液液萃取-GC-MS法测定水中硝基苯质量控制指标研究*
胡冠九,李娟,袁力 夏新
(国家环境保护地表水环境有机污染物监测分析重点实验室,江苏省环境监测中心,南京 210036) (中国环境监测总站,北京 100012)
采用液液萃取–气相色谱–质谱法测定水中硝基苯,通过统计全国多家实验室的测定数据,对平行样测定结果相对偏差、空白加标回收率、样品加标回收率、空白加标回收率相对偏差及样品加标回收率相对偏差5个质控指标进行分析,得出质控指标评价标准。在概率P,γ均为0.90时,平行样测定结果允许最大相对偏差应控制在11.0%;当空白加标浓度为0.2~30 μg/L时,回收率控制范围为59%~113%;当样品未检出、加标浓度在0.25~50 μg/L时,样品加标回收率控制范围为56%~110%;空白加标、样品加标回收率最大相对偏差应分别控制在10.0%和11.1%。在概率P和γ均为0.95时,平行样测定结果允许最大相对偏差应控制在13.5%;当空白加标浓度为0.2~30 μg/L时,回收率的控制范围为50%~122%;当样品未检出、加标浓度在0.25~50 μg/L时,样品加标回收率控制范围为49%~117%;空白加标、样品加标回收率最大相对偏差应分别控制在12.6% 和14.6%。
硝基苯; 质控指标;平行样测定结果相对偏差; 空白加标回收率; 样品加标回收率;空白加标回收率相对偏差;样品加标回收率相对偏差
环境污染物监测分析的结果是否准确可靠,应采取一定的质量保证和质量控制(QA/QC)措施进行全程序监控。从宏观上讲,一个检测机构的质量管理体系构建和运行、人员资质管理等均属于QA/QC内容;从微观上讲,每一批样品的分析质量如何,应有明确的控制指标及量化评价标准,用以判断此批样品的监测质量。因此质控指标的选择和评判标准的建立,对样品分析非常重要。
质控指标及其评价标准与所测定的污染物的性质、前处理方法、分析测试方法等密切相关。在我国现行的环境分析方法标准中,很少有专门的质控指标内容,有些方法仅在“注意事项”或“精密度和准确度”中对质控指标有所描述。以水中硝基苯分析方法为例,在GB/T 13194-91[1]中规定了“精密度”和“准确度”的指标,这是5个实验室对硝基苯类物质分别进行6次重复测定而获得的。在日常样品分析时(非样品考核、能力验证等),分析人员往往没有必要对同一水样进行3次以上的重复测定,也不太可能就一个样品,去寻找其它实验室进行平行测定。因此“精密度”和“准确度”这2个质控指标在日常分析工作中指导意义并不大,需要研究在日常工作中,实用、有效的质控指标及其评价标准。
笔者以硝基苯的监测为例,研究了实用的有机污染物监测质控指标,为其它有机污染的质控指标体系研究提供参考。
1 主流监测方法的确定
水中硝基苯测定方法主要有液液萃取-气相色谱分析法(GB/T 13194-91[1],HJ 592-2010[2],GB/T 5750.8-2006[3]),液液萃取/连续液液萃取-固相萃取-气相色谱-质谱法(EPA 8270c[4],EPA 625[5])、 固相萃取-气相色谱-质谱法(EPA 625[5],EPA 529[6])及高效液相色谱法(EPA 8330[7])。
对全国20个省的44家实验室(15个省级、29个地市级环境监测站)的调研结果表明,用液液萃取-气相色谱-质谱法分析硝基苯的共有19家,占45.2%,为主流分析方法。故笔者以液液萃取-GCMS为测定方法,利用其测定数据来建立质控指标体系。
2 质控指标研究内容
以实验人员在日常分析工作中“自我控制”为目的,建立相应的质控指标及评价体系,以期掌握日常批量数据的分析质量。主要研究液液萃取-GC-MS方法测定水中硝基苯时,平行样测定结果的相对偏差、空白加标回收率、实际样品加标回收率、空白加标回收率相对偏差及样品加标回收率相对偏差等5个质控指标的定量评价标准。
3 数据获得
用全国两级环境监测站(省站、市站)实验室在2005~2009年实际测定地表水获得的数据来计算质控指标。
具体监测数据来源见表1。

表1 质控指标计算的数据来源
4 异常值检验
对获得的监测数据进行统计检验,剔除异常值。统计检验的方法有:
(1)Dixon检验法:在剔除水平α=1%情况下,采用单侧检验,进行异常值判别[8]。除样品加标回收率测定值外,其它指标均用Dixon检验法检验。
(2)拉依达(Pauta)准则检验法:用于统计的实际样品加标回收率数据是各实验室不同浓度水平下的单个实测值,数据量较大,用拉依达(Pauta)准则来剔除异常值[9]。
5 结果与讨论
5.1 平行样测定结果的相对允许偏差
统计全国7个省的9家实验室对硝基苯的实际样品平行测定数据(笔者所述平行测定数量均为2,下同),其相对偏差的平均值(,%)为5.0,标准偏差(s,%)为2.8。假设平行样测定的实验室足够多,则平行样测定结果相对偏差应符合正态分布,所以将9家实验室的平行样测定结果相对偏差视为属于同一样本的9次测定,则不同概率P、概率γ下的k值及平行样相对偏差均值的高值单侧容许限详见表2。当γ为0.95,P为0.95时,查正态分布单侧容许限因子k(0.95,0.95,9)=3.031, 硝基苯平行样测定允许最大相对偏差+ks=13.5%,即有95%的把握认为,“平行样测定值相对偏差”单次值将会小于13.5%。当γ为0.90,P为0.90时,平行样测定允许最大相对偏差为11.0%。
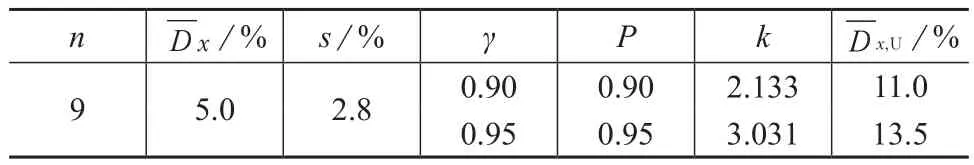
表2 9个实验室平行样相对偏差平均值的高值单侧容许限
本次调查的9个实验室中,8个实验室平行样相对偏差均值小于11.0%或13.5%,有1个实验室平行样相对偏差均值等于11.0%。
上述相对偏差的合理性可用以下几方面来进一步验证:全国12个省的13家实验室测定硝基苯时,12个实验室所测定的空白加标回收率的相对偏差均小于11.0%或13.5%;全国6个省的8家实验室测定硝基苯时,实际样品加标回收率相对偏差能够满足所规定的11.0%或13.5%。
5.2 空白样品的加标回收率及其相对偏差
12个实验室的空白加标浓度在0.2~30 μg/L区间内时,不同质控目标下的实验室空白加标回收率均值、空白加标回收率最低容许限和最高容许限见表3。
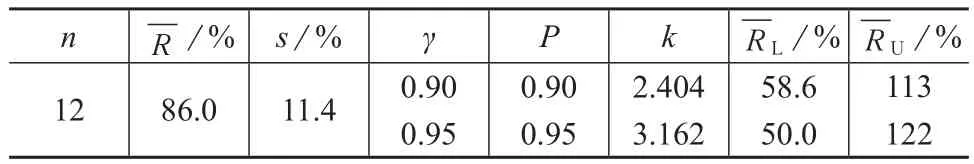
表3 12个实验室空白加标回收率均值的双侧容许限
黄敏等[11]对空白水样加标200 μg/L进行回收试验,得到测定结果的平均加标回收率为86%。解天民等[12]对空白水样加标40 μg/L进行回收率测定,得平均加标回收率为80%。刘良贵[13]对空白样品加标进行回收测定,得回收率为93.4%~106%。EPA 529方法[6]对空白水样分别加标0.1,5.0 μg/L进行回收试验,得平均加标回收率为70.6%~79.2%,87.8%~97.3%。上述文献报道值均在笔者所提出的允许范围之内。
全国11个省13家实验室对硝基苯的空白加标回收率相对偏差及不同概率P、概率γ平行样测定结果相对偏差均值的高值单侧容许限见表4。
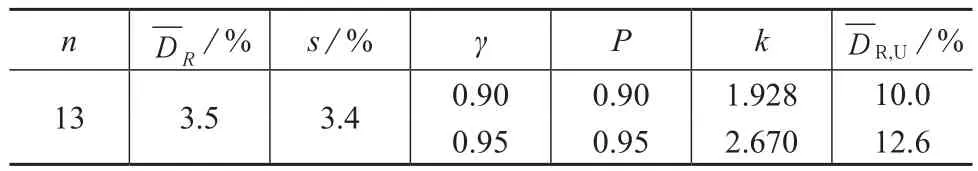
表4 13个实验室空白加标回收率相对偏差的高值单侧容许限
解天民等[12]对空白水样加标40 μg/L进行测定,得到回收率的相对偏差为0~3.1%。刘良贵[12]对空白样品分别加标3,15,30 μg/L进行平行样的测定并计算回收率,测定结果的相对偏差分别为1.0%~27.1%,0~10.7%和0.7%~8.75%。
5.3 实际样品的加标回收率及其相对偏差
34个实验室的样品测定浓度在未检出、加标浓度在0.32~150 μg/L区间内时,不同质控目标下的实验室样品加标回收率均值、样品加标回收率最低容许限和最高容许限见表5。
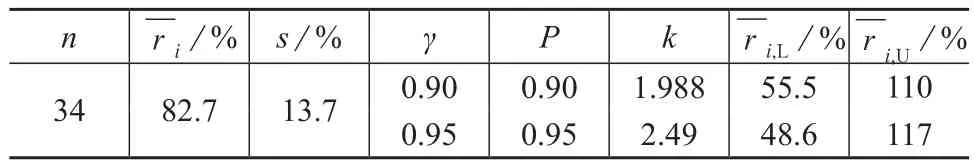
表5 34个实验室样品加标回收率均值的双侧容许限
EPA 8270c[4],EPA 625[5]多家实验室100 μg/L实际样品加标回收率统计结果表明,硝基苯的加标回收率可接受的控制范围为35%~180%。EPA 529方法[6]对实际水样加标5.0μg/L进行回收率的测定,测得平均加标回收率为88.0%~92.5%。
统计全国6个省的8家实验室对硝基苯的样品加标回收率相对偏差及不同概率P、概率γ平行样相对偏差均值的高值单侧容许限见表6。
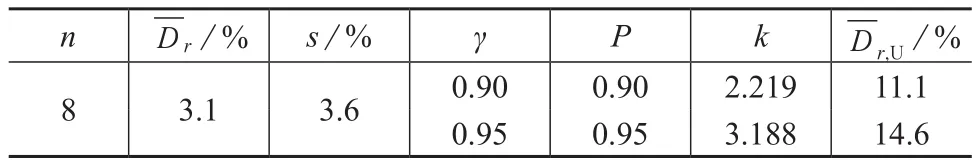
表6 8个实验室样品加标回收相对偏差的高值单侧容许限
6 结论
用液液萃取-气相色谱-质谱法测定水中硝基苯,当k和γ均为0.90时,平行样测定结果允许最大相对偏差为11.0%;k和γ均为0.95时,平行样测定结果允许最大相对偏差为13.5%。在空白加标浓度为0.2~30 μg/L,k和γ均为0.90时,空白加标回收率的范围为58.6%~113%,空白加标回收率的最大允许相对偏差为10.0%;k和γ均为0.95时,空白加标回收率的范围为50.0%~122%,空白加标回收率的最大允许相对偏差为12.6%。在样品未检出,加标浓度在0.25~50μg/L,k和γ均为0.90时,样品加标回收率范围为55.5%~110%,样品加标回收率最大允许相对偏差为11.1%;k和γ均为0.95时,实际样品加标回收率范围为48.6%~117%,样品加标回收率最大允许相对偏差为14.6%。
上述质控指标范围能够涵盖相关文献报道值,并有所放宽,能反映实际工作情况。
[1] GB/T 13194 – 1991 水质、硝基甲苯、硝基氯苯、二硝基甲苯的测定 气相色谱法[S].
[2] HJ 592–2010 水质 硝基苯类化合物的测定 气相色谱法[S].
[3] GB/T 5750.8 – 2006 生活饮用水标准检验方法 有机物指标[S].
[4] EPA 8270c 气相色谱/质谱法测定半挥发性有机物[S].
[5] EPA 625 GC/MS法测定工业废水水中部分有机化合物[S].
[6] EPA 529 固相萃取-气相色谱/质谱法测定饮用水中爆炸物及相关化合物[S].
[7] EPA 833 高效液相色谱法测定硝基苯类和硝胺类化合物[S].
[8] 中国环境监测总站《环境水质监测质量保证手册》编写组.环境水质监测质量保证手册[M].北京:化学工业出版社,1994.
[9] 张敏,袁辉.拉依达(PauTa )准则与异常值剔除[J].郑州工业大学学报,1997,18(1): 84–88.
[10] 蒋子刚,顾雪梅.分析检验的质量保证和计量认证[J].上海:华东理工大学出版社,1998: 56 .
[11] 黄敏,唐莺,沈咏洁,等.GC-MS/SIM法测定地表水中半挥发性有机化合物[J].净水技术,2008,27(2): 66–69.
[12] 解天民.环境分析化学实验室技术与运营管理[M].北京:中国环境科学出版社,2008.
[13] 刘良贵.气相色谱法测定水中硝基苯及一二、三硝基甲苯[J].国土资源,1989(3): 269–277.
Research on Quality Control Index for Determination of Nitrobenzene in Water by GC-MS After liquid-liquid Extraction
Hu Guanjiu, Li Juan, Yuan Li Xia Xin
(Jiangsu Environmental Monitoring Center, Nanjing 210036, China) (China Environmental Monitoring Station, Beijing 100012, China)
Five indexes of quality control for determination of nitrobenzene by GC-MS after liguid-liquid extraction were given by evaluating datas collected from a few labs national-widely, including the relative deviation of the duplicates detection results, the recoveries and the relative deviation of the blank sample with standard addition, the recoveries and the relative deviation of the real sample with standard addition. The quality control indexes were produced as the probability of both P and γ were 0.90, the maximum relative deviation of the detection results should be within 11.0%; the blank recoveries should be within 59%-113% as the addition concentration was 0.2-30 μg/L; the sample recoveries should be within 56%-110% when the nitrobenzene was not detected in all the samples and the addition concentration was 0.25-50 μg/L; the maximum relative deviation of the blank sample with standard addition should be within 10.0%, and the maximum relative deviation of sample with standard addition should be within 11.1%. When the probability of both P and γ were 0.95, the maximum relative deviation should be within 13.5%; the blank recoveries should be within 50%-122% as the addition concentration was 0.2-30 μg/L; the sample recoveries should be within 50%-122% as the nitrobenzene was not detected in all the samples and the addition concentration was 0.25-50 μg/L; the maximum relative deviation of the blank sample with standard addition should be within 12.6%, and the maximum relative deviation of the sample with standard addition should be within 14.6%.
halogenated hydrocarbon; quality control index; relative deviation of duplicates; blank recovery; sample recovery; relative deviation of blank sample with standard addition; relative deviation of sample with standard addition
O657.7
A
1008–6145(2012)02–0025–04
10.3969/j.issn.1008–6145.2012.02.007
* 环保公益性行业科研专项(200809140)
联系人:胡冠九; E-mail: huguanjiu@163.com
2011–12–22
猜你喜欢
四川大学学报(自然科学版)(2025年1期)2025-02-10 00:00:00
食品安全导刊(2021年21期)2021-08-30 08:21:48
理化检验-化学分册(2020年12期)2021-01-26 00:41:32
山西大同大学学报(自然科学版)(2016年6期)2016-01-30 08:29:27
分析测试学报(2015年6期)2016-01-13 06:18:58
分析测试学报(2015年5期)2016-01-13 06:18:52
岩矿测试(2015年3期)2015-12-21 03:57:04
核科学与工程(2015年2期)2015-09-26 11:57:23
应用化工(2014年4期)2014-08-16 13:23:09
中国氯碱(2014年10期)2014-02-28 01:04:59
