
中国浓香型白酒窖池糟醅中微生物群落演替分析
2012-06-01 09:25张大凤刘若尘邢亚阁李明元车振明向文良
食品科学 2012年15期
张大凤,李 可,刘 森,刘若尘,邢亚阁,李明元,车振明,向文良,*
(1.西华大学生物工程学院 四川省食品生物技术重点实验室,四川 成都 610039;2.西华大学古法发酵(酿造)生物技术研究所,四川 成都 610039)
中国浓香型白酒窖池糟醅中微生物群落演替分析
张大凤1,2,李 可1,2,刘 森1,2,刘若尘1,邢亚阁1,李明元1,2,车振明1,向文良1,2,*
(1.西华大学生物工程学院 四川省食品生物技术重点实验室,四川 成都 610039;2.西华大学古法发酵(酿造)生物技术研究所,四川 成都 610039)
为探索中国浓香型白酒发酵糟醅中微生物群落演替规律,利用克隆分析,从时间和空间两个层面对糟醅微生物区系的消长变化进行较为全面的调查。根据有效克隆子分布数据计算的群落多样性参数表明:窖池糟醅微生物群落多样性在时间和空间上存在差异。应用CANOCO 4.5 软件对物种和生态因子进行典范对应分析(CCA),应用CANODRAW 4.0作种类、样方分布与生态因子关系的二维排序图,排序图上清楚地反映了窖池糟醅微生物种类、群落分布与生态因子的关系。结果表明:酸度、乙醇含量、水分含量是影响微生物群落结构组成的主要微生态因子;大部分微生物适宜在低酸度条件下生长,少数耐酸微生物能在高酸度条件下生长;随着发酵的进行,生态因子发生变化,微生物群落也进行相应的演替,向耐高酒精含量、高酸度的群落发展。
中国浓香型白酒;发酵糟醅;微生物演替
中国浓香型白酒的生产以泥窖窖池为基础,发酵过程是栖息在窖池糟醅、窖泥中的庞大微生物区系在糟醅固、液、气三相界面的复杂的物质能量代谢过程[1-2]。在这一过程中,窖池生态因子如窖池温度、糟醅酸度、总糖及还原糖含量、糟醅水分含量等与窖池中的微生物彼此联系、互相促进、互相制约、协调发展,共同构成了窖池特定的微生物生态系统,并在“三流运转”规律的交互作用下构成了发酵过程中窖池微生态的动态发展和特定阶段的动态平衡[3-4]。由于环境信息的传递对于调控窖池微生物种类的变化、代谢产物的生成与积累至关重要,并决定着窖池物质代谢和能量代谢的走向。因此,环境生态因子的变化对微生物的生长、代谢影响非常大。在窖池不同空间位置和微生物生长代谢的不同阶段,微生物对窖池生态因子的需求都有所不同[5]。随着发酵的进行,微生物的演替活动将引起窖池环境生态因子不断发生变化,而这些变化又将反过来作用于窖池微生物,从而影响窖池发酵,最终导致窖池中各代谢产物成分和比例的变化,从而影响窖池浓香型白酒的产量及品质[6-7]。所以,研究窖池微生物的动态变化对于把握窖池发酵情况是很有必要。正因如此,白酒窖池生态系统中微生物的演替引起了广大学者的极大兴趣,但系统分析其演替过程,还未见报道。
本研究从生态学角度审视传统固态发酵,利用生态分析相关软件,对中国白酒窖池糟醅中微生物群落和生态因子等相关数据进行数理统计,以揭示伴随发酵进行,窖池不同空间层次糟醅中微生物群落物种多样性变化趋势和演替规律以及在窖池不同空间层次和不同发酵时间的糟醅中微生物群落结构与窖池微生态因子之间的关系,寻找对糟醅微生物群落结构起关键作用的生态因子,探讨影响微生物群落演替的机制,为理解中国浓香型白酒发酵机理、特征风味形成机制提供参考。
1 材料与方法
1.1 数据采集
分析所用的微生物群落和糟醅生态因子数据均由西华大学古法发酵(酿造)生物技术研究所提供。样品采集于四川某著名浓香型白酒企业48号窖池,分别为窖池上、中、下层中心发酵时间1、7、6 0 d的糟醅。其中,上层糟醅样分别记为:T01、T07、T60;中层糟醅样分别记为:M01、M07、M60;下层糟醅样分别记为:B01、B07、B60。所有样品中的pH值、酸度、还原糖、总糖、乙醇、温度、水分、乙醛、酯和蛋白质等微生态因子依据沈怡方等[8]的相关方法测定。糟醅中微生物群落的分析参考向文良[7]、Zhang Wenxue[9]等的方法,PCR分别扩增原核和真核微生物的16S rRNA基因和18S rRNA基因。扩增的16S rRNA基因和18S rRNA基因再分别与pGM-T载体连接成重组质粒后,分别导入感受态的E.coli DH5α构建窖池糟醅中原核微生物的16S rRNA基因文库和真核微生物的18S rRNA基因文库,提取重组质粒,基因测序分析窖池糟醅中微生物的分类学地位。窖池糟醅的生态因子数据见表1。窖池糟醅中微生物群落数据见表2,其中原核微生物代表克隆子的16S rRNA基因在GenBank(http://www. ncbi.nlm.nih.gov/genbank/)中的接受号:JQ693078~JQ693100;真核微生物代表克隆子的18S rRNA基因在GenBank中的接受号:JQ693061~JQ693077。
1.2 数据分析方法
微生物群落关系的演替关系利用Origin8.0绘制。群落多样性参数Simpson指数(D)、Shannon-Wiener多样性指数(H)、均匀度(J)的计算公式分别如下:

式中:pi为第i种物种个体数占群落总个体数的比例;S为物种总数;Hmax为H的最大值,等于lnS[10]。Shannon-Wiener 多样性指数随时间的动态变化图用SPSS for Windows 13.0绘制。窖池糟醅中微生物群落与环境的相互作用关系利用典范对应分析(canonical correspondence analysis, CCA)排序[11]。

表1 窖池糟醅的生态因子Table 1 Chemical composition of lees (wet weight basis)
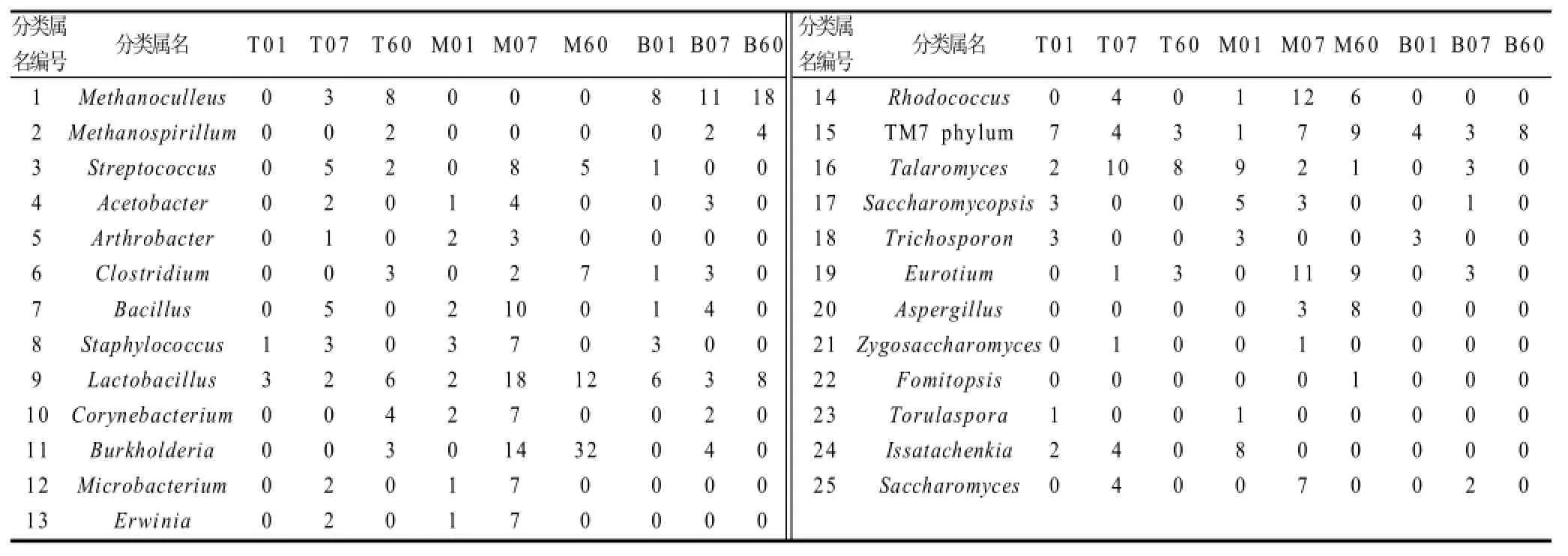
表2 窖池糟醅样中微生物16S rRNA和18S rRNA基因的克隆分布Table 2 Microbial categorization and abundance in fermentation lees based on the 16S rRNA and 18S rRNA analysis
2 结果与分析
2.116 S rRNA和18S rRNA基因的克
隆分析
2.1.1 上层糟醅中微生物分析
基于16S rRNA和18S rRNA分析技术表明:入窖样获得了22个有效克隆,发酵7d样获得了53个有效克隆,发酵60d样获得了42个有效克隆,这些克隆子代表的微生物伴随发酵时间发生的群落演替如图1所示。上层糟醅对原核微生物而言,入窖样中未检出古细菌,发酵7d样和6 0 d样检出的古菌为M e t h a n o c u l l e u s属和Methanospirillum属;其中Methanospirillum属只在发酵60d样被检出。入窖样中,细菌、霉菌、酵母的种类和数量差异不是很大。随着发酵进行和糟醅理化因子的变化,糟醅中微生物群落也发生明显变化,特别是细菌,其种类和数量发生明显变化(图2)。入窖样中未出现的Streptococcus 属、Acetobacter属、Arthrobacter属、Bacillus属、Microbacterium属、Erwinia属和Rhodococcus属均在发酵7d样中检出。在60d样中,已检测不到酵母了,因为这时发酵已结束,此时窖池环境恶劣,如乙醇含量度高、严格厌氧、酸度高,酵母无法生存[12-13],此时在60d样品中只有甲烷菌、霉菌、细菌。

图1 窖池上层糟醅中微生物16S rRNA和18S rRNA基因的克隆分布图Fig.1 Microbial categorization and abundance in the top layer of fermented lees based on the 16S rRNA and 18S rRNA analysis
2.1.2 中层糟醅中微生物分析
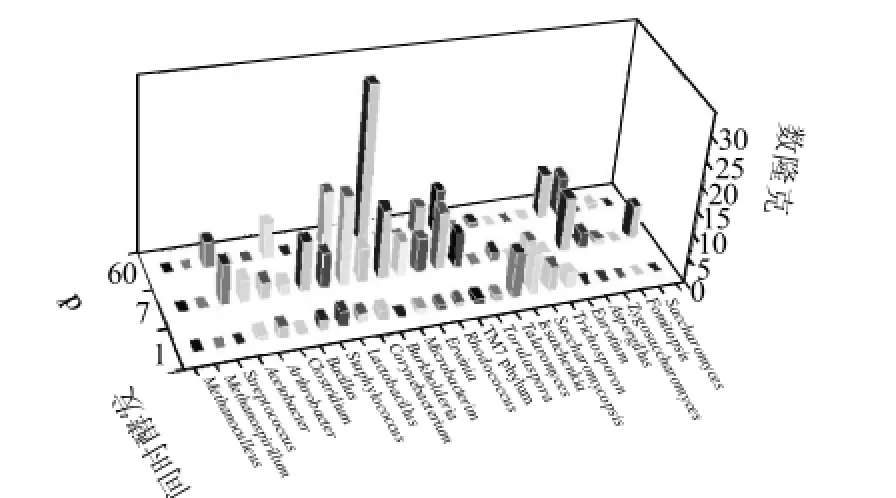
图2 窖池中层糟醅中微生物16S rRNA和18S rRNA基因的克隆分布图Fig.2 Microbial categorization and abundance in the middle layer of fermented lees based on the 16S rRNA and 18S rRNA analysis
中层样主要是母糟,包括红糟和粮糟,是发酵最活跃的区域[14],微生物的种类和数量在此糟醅中发生剧烈演替。由图2可知,入窖样获得了42个有效克隆,发酵7d样获得了133个有效克隆,发酵60d样获得了90个有效克隆,其数量明显多于上层样和下层样。上层中的Methanoculleus属和Methanospirillum属在中层中没有检出,其他古菌也未检出。Burkholderia属在上层样中演替并不活跃,但中层糟醅中尽管入窖时中未检出,但是当其在发酵7d时Burkholderia属的克隆数迅速达到14个,在发酵60d时其进一步上升,达到32个克隆,占发酵60d总克隆数的35.6%,此时Burkholderia属成为优势属。Talaromyces属在上层样中比较活跃,一度成为上层发酵7d糟醅中的优势菌。在中层糟醅中随着发酵的进行,Talaromyces属中微生物数量一直递减,在发酵7d样和60d样中分别只能检测出2个和1个克隆子。Eurotium 属、Aspergillus属在中层中含量也比较丰富,这与中层样中含有的粮糟有关。同样,在中层糟醅发酵60d时也未检出酵母。
2.1.3 下层糟醅中微生物分析
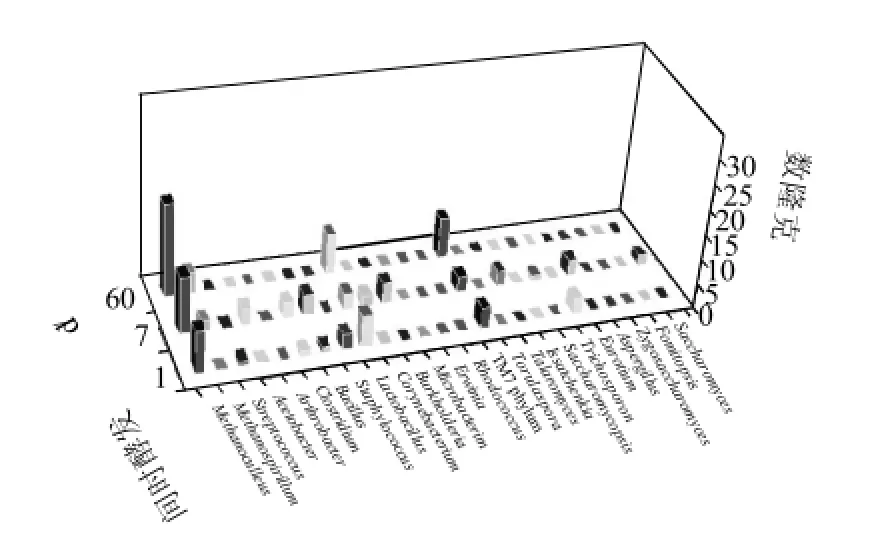
图3 窖池下层糟醅中微生物16S rRNA和18S rRNA基因的克隆分布图Fig.3 Microbial categorization and abundance in the bottom layer of fermented lees based on the 16S rRNA and 18S rRNA analysis
由图3可知,在下层糟醅中入窖样获得了27个有效克隆,发酵7d样获得了44个有效克隆,发酵60d样获得了38个有效克隆。下层样是多轮底糟,其酸度非常高,所以微生物种类和数量都相对较少,在发酵过程中微生物种类和数量的波动比较平缓。下层样另一特点是古菌含量丰富,包括甲烷菌M e t h a n o c u l l e u s属和Methanospirillum属,入窖糟醅中古菌占总菌属的29.6%,7d为29.5%,60d为57.9%。发酵结束后,只有几种耐酸性的细菌存在,主要为细菌中的Lactobacillus属和TM7 phylum;霉菌和酵母在此时未被检出。
2.2 窖池中微生物群落多样性分析

图4 Shannon-Wiener多样性指数随发酵时间的变化Fig.4 Temporal changes of microbial Shannon-Wiener diversity
窖池糟醅上、中、下层的入窖样、发酵7d样、发酵60d样共9个样方中分布的微生物菌群多样性分析结果见图4。Shannon-Wiener多样性指数在上、中、下层糟醅中表现出同样的变化趋势:随着发酵的开始,群落多样性增加,到发酵结束时,群落多样性降低。发酵开始时,糟醅原料提供了丰富的营养物质和适宜的生长条件,各类微生物能快速生长。随着氧气和营养物质的消耗,代谢产物的积累,窖池环境变得越来越不适宜微生物的生长。下层样由于酸度最高,微生物种类最少,主要是一些耐酸的微生物能生长,故其Shannon-Wiener多样性指数在入窖样、发酵7d样、发酵60d样中均低于上层样和中层样。中层样是发酵最活跃的区域,也是微生物种类和数量最丰富的区域,其多样性指数在入窖样和发酵7d样中均大于上层样和下层样。但中层样在发酵60d样中却低于上层样,可能是由于中层样与下层样间通过黄水的物质交换比较频繁,中层发酵60d样中的酸度也比较高,导致微生物多样性低于上层样。
2.3 微生物群落演替与生态因子之间的关系
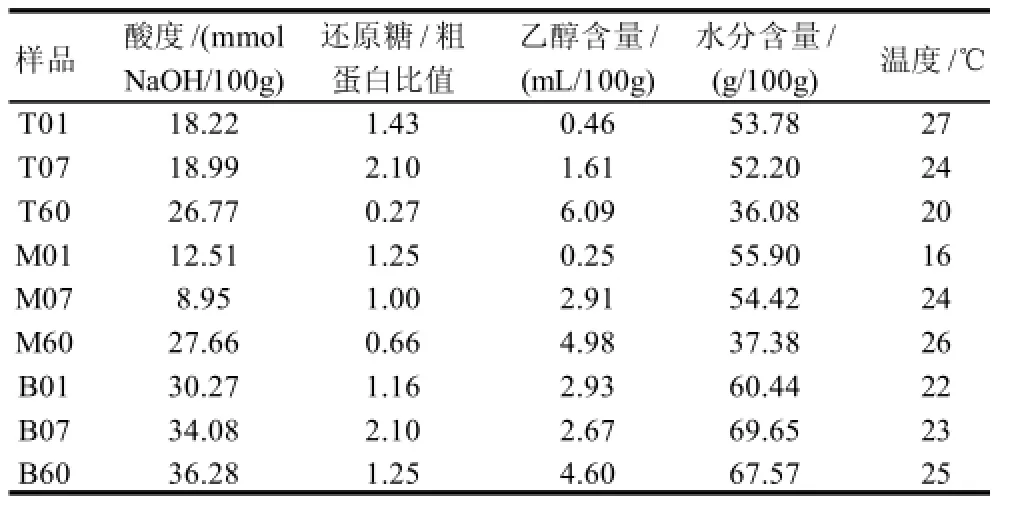
表3 窖池糟醅的微生态因子Table 3 Microecological factors in fermentation less

表4 CCA分析的统计信息Table 4 Statistical analysis of CCA analysis results
9个样方的生态因子经过处理后(表3)与微生物的种类用于CCA排序。CCA排序结果(表4)表明:轴1和轴2两个排序轴的本征值分别为0.398和0.296;生态因子与微生物种类相关性分别为0.998和0.868;物种数据的方差累计百分数分别为30.7和53.6;总本征值为1.294,总典范本征值为1.036,解释量为80.06%;蒙特卡罗检验值小于等于0.05,所以排序结果较好的反映了微生物种类组成与生态因子间的相互关系[15-16]。从CCA排序图来看,酸度梯度是所有因子中对微生物群落分布起决定性作用的生态因子,大多数物种都分布在低酸度区域,如图5所示。低酸度区域的物种多样性远远高于高酸度区域[17]。Methanoculleus(1)和Methanospirillum(2)属对酸度的要求高,是糟醅中最耐酸的微生物,且对水分的要求也高。TM7 phylum(15)、Lactobacillus(9)和Clostridium(6)属也能耐一定的酸度。Trichosporon(18)、Saccharomycopsis(17)、Torulaspora(23)和Issatchenkia(24)属主要分布在还原糖/粗蛋白质比值高、水分含量较高、乙醇含量低、低酸度、相对温度较低的区域,其中Tri-chosporon(18)属对水分的要求较高 。Talaromyces(16)、Staphylococcus(8)、Arthrobacter(5)、Acetobacter(4)、Bacillus (7)和Corynebacterium(10)属,多见于具有适宜的水分含量、合适的还原糖/粗蛋白质和低酸度的区域。Sa ccharomyces(25)、Microbacterium(12)、Erwinia(13)、Zygosaccharomyces(21)、Rhodococcus(14)和Streptococcus (3)属,主要分布在适宜在水分含量低、温度较高、酸度低和一定含量酒精的糟醅中。Fomitopsis(22)、Aspergillus(20)、Burkholderia(11)、Eurotium(19)和Clostridium(6)属的微生物与糟醅乙醇含量有着极大地相关性,在较高乙醇含量条件下也能生存。

图5 微生物物种与生态因子的CCA二维排序Fig.5 Canonical correspondence analysis based on microbial species and ecological factors
3 结 论
3.1 统计分析表明,在发酵不同时间、窖池糟醅不同空间层次中微生物类群和数量存在较大的差异,以中层样居首。
3.2 通过对多样性分析表明,窖池糟醅微生物群落多样性随空间和发酵时间的变化而异,具体表现为:入窖样和发酵7d样的群落多样性分别为中层>上层>下层,发酵60d样的群落多样性为上层>中层>下层。在时间轴上,糟醅微生物群落多样性呈先上升后下降的趋势。
3.3 CCA排序结果显示:酸度、乙醇含量、水分含量是影响微生物群落结构组成的主要微生态因子。除了极少数耐酸的微生物,如甲烷菌,Lactobacillus属能在较高的酸度下生存,大部分微生物生活在低酸度区域。随着发酵的进行,乙醇逐渐积累,微生物群落发生相应的演替,向耐高酒精含量、高酸度的群落发展。
[1]张文学, 乔宗伟, 向文良, 等. 中国浓香型白酒窖池微生物生态研究进展[J]. 酿酒, 2004, 31(2): 31-35.
[2]赵发清, 马海燕. 微生态基本概念的剖析[J]. 中国微生态学杂志, 1997, 7(6): 41-44.
[3]罗海, 刘森, 向文良, 等. 浓香型白酒窖池微生物生态因子的动力学分析[J]. 中国酿造, 2011(7): 108-112.
[4]唐由凯. 论微生态系统及系统分析[J]. 中国微生态学杂志, 2000, 12 (2): 116-118.
[5]张文学. PCR技术对浓香型白酒糟醅细菌菌群的解析[J]. 四川大学学报: 工程科学版, 2005, 37(5): 82-87.
[6]王大珍. 微生物生态学的发展及应用[J]. 科学, 1993(2): 18-20.
[7]向文良, 乔宗伟, 张文学, 等. 中国浓香型白酒窖池中原核微生物的特性及系统发育分析[J]. 四川大学学报: 工程科学版, 2005, 37(1): 39-42.
[8]沈怡方, 高景炎, 康明官, 等. 白酒生产技术大全[M]. 北京: 中国轻工业出版社, 2007: 597-671.
[9]ZHANG Wenxue, QIAO Zongwei, TANG Yueqin, et al. Analysis of the fungal community in Zaopei during the production of Chinese Luzhou flavour liquor[J]. Journal of the Institute of Brewing, 2007, 113(1): 21-27.
[10]KEYLOCK C J. Simpson diversity and the Shannon-Wiener index as special cases of a generalized entropy[J]. Oikos, 2005, 109(1): 203-207.
[11]TERAHARA T, IKEDAB S, NORITAKE C, et al. Molecular diversity of bacterial chitinases in arable soils and the effects of environmental factors on the chitinolytic bacterial community[J]. Soil Biol Biochem, 2009, 41(3): 473-480.
[12]向文良. 中国浓香型白酒窖池微生物生态研究[D]. 成都: 四川大学, 2004.
[13]施安辉. 浓香型白酒发酵过程窖中微生物区系的分析[J]. 酿酒, 1986 (4): 24-29.
[14]吴衍庸. 白酒工业生态中的微生物生态学[J]. 酿酒科技, 2001(5): 32-33.
[15]张金屯. 数量生态学[M]. 北京: 科学出版社, 2003: 131-193.
[16]郝占庆. 长白山北坡木本植物分布与环境关系的典范对应分析[J]. 植物生态学报, 2003, 27(6): 733-741.
[17]MACDONALD C A, THOMAS N, ROBINSON L, et al. Physiological, biochemical and molecular responses of the soil microbial community after afforestation of pastures with Pinus radiata[J]. Soil Biol Biochem, 2009, 41(8): 1642-1651.
Microbial Community Succession of Chinese Luzhou-Flavor Liquor Lees
ZHANG Da-feng1,2,LI Ke1,2,LIU Sen1,2,LIU Ruo-chen1,XING Ya-ge1,LI Ming-yuan1,2,CHE Zhen-ming1,XIANG Wen-liang1,2,*
(1. Provincial Key Laboratory of Food Biotechnology of Sichuan, College of Bioengineering, Xihua University, Chengdu 610039, China;2. Biotechnology Institute of Ancient Brewing, Xihua University, Chengdu 610039, China)
In order to explore the succession of microorganism communities in fermented grains from Chinese Luzhou-flavor liquor production, we investigated the temporal and spatial changes of microbial communities by clone analysis. The community diversity parameters calculated based on clone distribution suggested that microbial community diversity showed temporal and spatial variations. The data of species and ecological factors were analyzed by canonical correspondence analysis (CCA) using CANOCO 4.5. The species (or sites)-environment biplot of CCA were automatically mapped using CANODRAW 4.0 and the relationships of the ecological distribution of the fermented grains microorganism species and communities with the ecological factors were clearly revealed on these biplots. The results showed that the acidity, ethanol and humidity had important influences on the microbial community composition. The bulk of microorganism species adapted to grow in low acidity condition and only a few microorganisms could live in high acidity condition. The microorganism communities responded to changes of ecological factors along with fermentation, towards a community tolerant of high levels of ethanol and acidity.
Chinese Luzhou-flavor liquor;fermentation lees;community succession
TS261.1
A
1002-6630(2012)15-0183-05
2011-11-11
教育部春晖计划项目(Z2010101);四川省食品生物技术重点实验室开放基金项目(SZJJ2009-014);
西华大学重点科研基金项目(Z0910501)
张大凤(1973—),女,讲师,硕士研究生,研究方向为传统发酵食品生物技术。E-mail:yhf1998914@163.com
*通信作者:向文良(1973—),男,副教授,博士,研究方向为传统发酵食品微生物过程学。E-mail:biounicom@mail.edu.cn
猜你喜欢
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09
农村百事通(2020年22期)2020-12-28
酿酒科技(2020年1期)2020-04-30
世界热带农业信息(2018年10期)2018-03-14
现代园艺(2017年23期)2018-01-18
数学杂志(2017年3期)2017-06-15
少年文艺·开心阅读作文(2017年1期)2017-02-24
考古与文物(2016年5期)2016-12-21
浙江农业科学(2016年11期)2016-05-04
小天使·四年级语数英综合(2014年3期)2014-03-21
