
氧化铈对水煤气变换反应催化剂的影响研究
2012-05-15 08:08樊惠玲李晓东上官炬林建英郭汉贤
太原理工大学学报 2012年1期
樊惠玲,李晓东,上官炬,林建英,郭汉贤
(太原理工大学 煤科学与技术省部共建国家重点实验室培育基地,太原030024)
水煤气变换反应在合成氨工业中一直占有重要地位。近年来,燃料电池等洁净氢能源日渐受到重视。利用变换反应将富氢燃料中对阳极催化剂有害的CO转化为H2,不仅可降低CO的含量,同时也可有效提高H2的含量,已成为制氢的重要技术途径之一[1-2]。由此可看出变换反应在化工能源领域的重要作用。传统的中温变换催化剂为铁铬系催化剂,其在合成氨工业上应用已有多年历史,至今在国内市场仍占有较高份额。在变换催化剂中,氧化铬的质量分数一般约在8%左右[3],作为结构助剂,用以提高活性及耐热性[4-5]。然而,氧化铬不仅价格昂贵,且对环境会造成严重污染。因此,开发研制具有良好活性又环境友好的无毒价廉的铁系低铬或无铬变换催化剂势在必行。
稀土元素可提供良好的电子转移轨道,其中铈有变价性、贮氧性能以及良好的氧化还原特性,在许多的催化反应中已得到应用[1,6,7]。以铈部分或全部代铬的改进高变催化剂也相继出现[8-12]。如,内蒙古工业大学研制的BX型高变催化剂,其中Cr2O3质量分数仅为3%,CeO2质量分数为2%[11]。本研究组曾对不同含量的含铈催化剂进行了程序升温还原(TPR)的研究,并对CeO2的助剂作用进行了一定探讨[13-14]。本论文主要报道采用程序升温氧化(TPO)与脱附(TPD)技术研究氧化铈含量对中变催化剂的影响结果。
1 实验部分
1.1 催化剂的制备
实验所用催化剂样品由干混法制备,样品主要组成及含量见表1。
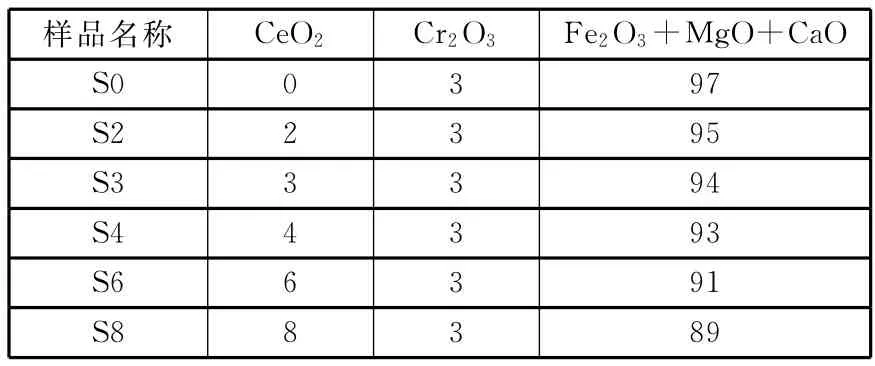
表1 催化剂样品组成(质量分数) %
1.2 催化剂的程序升温技术表征
1.2.1 样品预处理
为了了解变换反应的过程机理,采用程序升温技术对变换反应所涉及的反应物与产物,即H2、CO、H2O及CO2进行了实验研究。需注意要先排除再吸附和内扩散因素的影响,使实验在动力学区进行。样品在使用前先经粉碎,并在马弗炉中于350℃下焙烧2h。本研究在预实验基础上选择催化剂粒度40~60目,样重0.3g。
催化剂在TPO及TPD吸附气体之前,要先经过还原。将样品装入反应管中,升温至310℃并用氩气吹扫1.5h,之后切换进入H2还原2h,将催化剂还原为Fe3O4状态。还原结束后,切换进氩气,升温至380℃,吹扫至基线平稳,然后降温至200℃,留待程序升温实验使用。
1.2.2 程序升温氧化 H2O-TPO
催化剂样品进行如上还原处理后,切入H2O-Ar混合气,以H2O作为氧化介质进行TPO研究。出口气体用冷阱除水后,进入热导池检测臂检测。升温速率10℃/m in,所用载气Ar流量为40mL/min。
1.2.3 程序升温脱附CO/CO2-TPD
催化剂样品在经如上还原预处理后,在200℃切入CO或CO2经一定时间吸附饱和后,再切换进氩气吹扫,至色谱基线平稳,以10℃/min的速率开始升温脱附。
2 结果与讨论
2.1 H2O-TPO研究
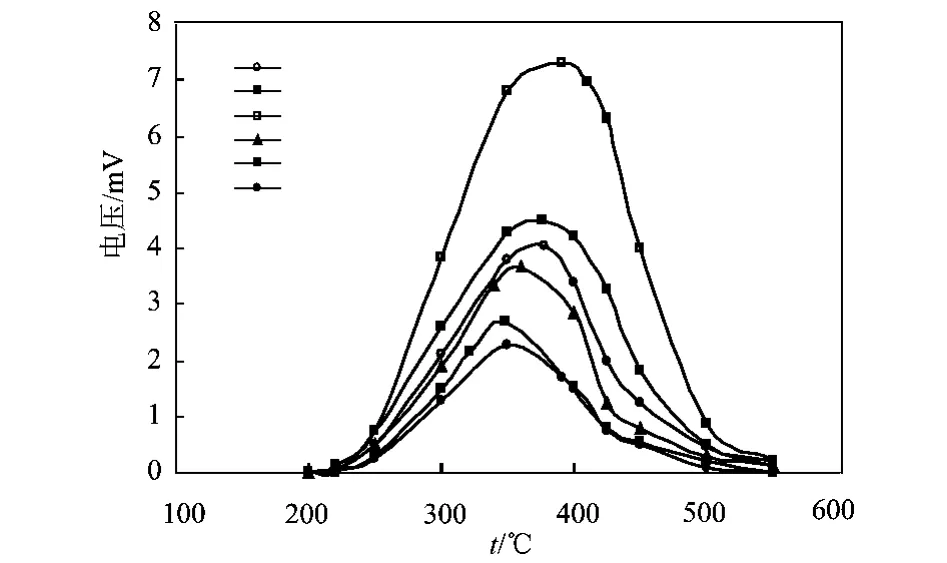
图1 各催化剂的TPO图
采用Ar+H2O的混合气氛,H2O作为氧化介质,研究CeO2含量对还原样品的TPO结果的影响。在程序升温氧化过程中,随着温度升高,吸附态的H2O使样品发生氧化,自身则被还原为氢气。因此,TPO所得到的谱图实为产氢量的信息随温度的变化情况,如图1所示。由图可见,还原样品的起始氧化温度约为240℃;随着CeO2含量的增加,TPO的峰温增高,峰面积也增大,并在CeO2质量分数为3%时达到极值。之后,峰温与峰面积均随着CeO2含量的增加而减低。各样品TPO所对应的峰面积及产氢量见表2。将样品产氢量与其CeO2含量进行关联,见图2。明显看出,CeO2对氧化过程的改善以样品S3最为显著。
对比TPO与TPR[13]的结果,发现TPR的tm在400~410℃之间,而TPOtm在360~390℃之间,峰温降低说明氧化容易进行。这是因为CeO2促进Fe2+失去电子而被氧化。梁斌[15]通过XPS测定认为,Ce的加入使得Fe 2p3/2电子结合能降低,从而使氧化变得容易。
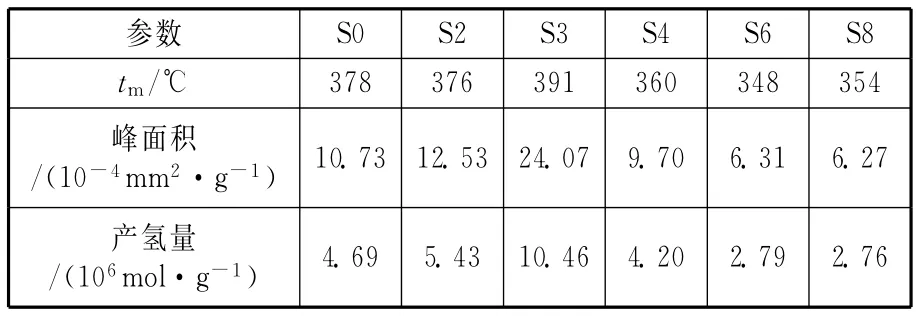
表2 各催化剂样品的TPO谱图参数
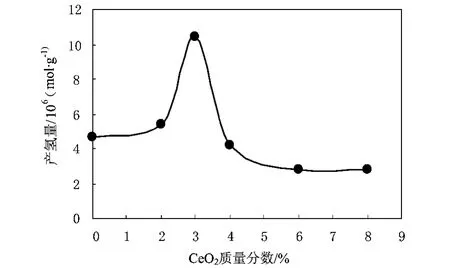
图2 不同CeO2含量的催化剂样品与其产氢量的关系
另外,TPR的第一峰对应Fe2O3至Fe3O4的还原,将这一过程的耗氢量[13]与TPO的产氢量相比,发现TPO的产氢量小于TPR的耗氢量。这说明,H2O的氧化作用并不能使由还原过程产生的Fe3O4完全返回至Fe2O3,只能回到Fe3+与Fe2+之间的某个中间价态。这可以从两方面说明。首先,本研究TPO采用的氧化剂为H2O而不是O2,H2O的氧化能力显然不及O2。其次,从气固反应的特性来讲,样品还原是其中氧逐渐减少过程,也即是个开孔的体相过程。反过来,样品从还原态到氧化态则是个闭孔过程。因此,随反应进行,样品体相内部的氧化反应相对还原较难进行,限制了H2O的氧化向样品深度发展。以上两点原因使得H2O在有限的时间内不足以将样品中Fe3O4完全氧化到Fe2O3的程度,氧化反应因此出现滞后的现象。
2.2 CO-TPD研究
图3为催化剂S6的CO-TPD谱图,图中两条曲线分别对应于5min和13min的不同吸附时间。谱图各点正比于相应温度下的脱附速率,峰值则对应于最大脱附速率。其它几种催化剂的CO-TPD谱图与此相似,不再赘述。将各催化剂的峰值与CeO2含量作图,见图4。由TPD谱图可计算出相应的CO脱附量,见表3。可见,在两种吸附时间下,CeO2质量分数为3%的样品S3对CO的吸附能力最好。这可能与催化剂表面酸碱的变化有关。
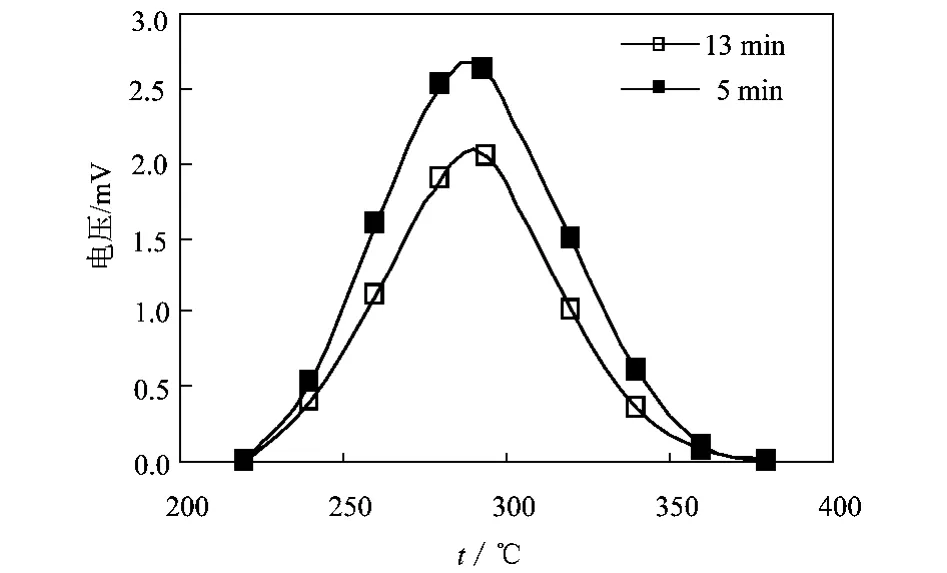
图3 样品S6的CO-TPD图
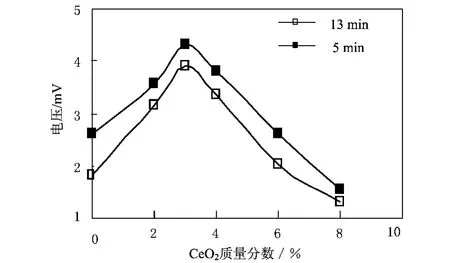
图4 不同CeO2含量的样品与其CO脱附量的关系
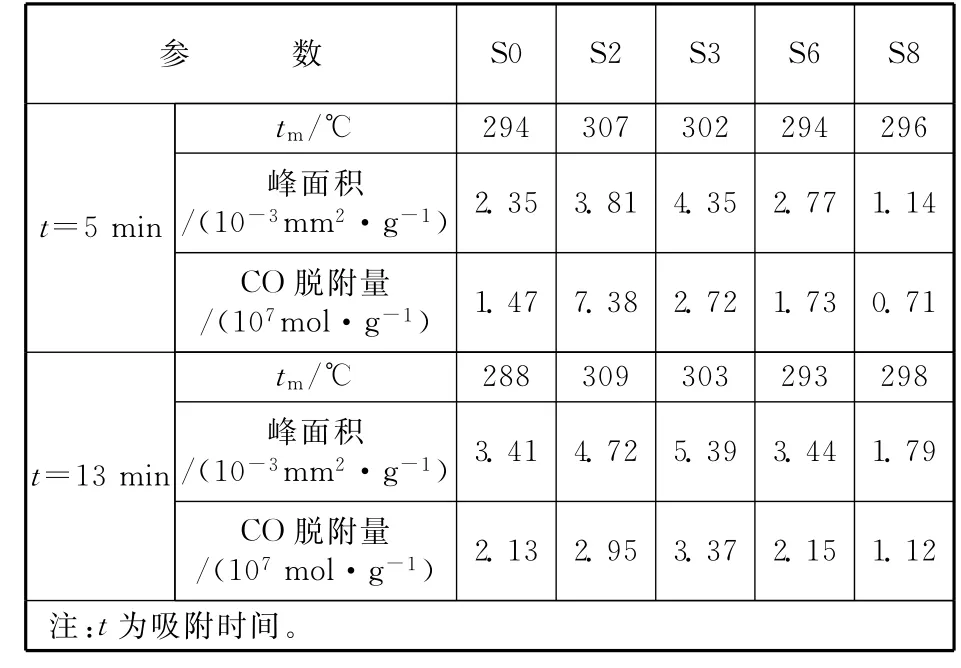
表3 各催化剂的CO-TPD参数
对中变Fe基催化剂的催化反应机理主要有两种解释,即氧化还原与甲酸盐中间物机理[6]。但无论哪种机理,CO在催化剂表面的吸附是必不可少的一步。普遍认为,CO在变换催化剂上的吸附主要是与铁离子的配位吸附。少量氧化铈的加入可以提高活性组分的分散度,增大比表面积,因而也利于CO吸附。但由于Ce离子与Fe离子的离子半径相差较大,Ce离子不易进入Fe3O4晶体内部形成固溶体。虽然少量加入会引起晶格畸变,但加入量过大时,氧化铈本身易积聚,并堆积在催化剂表面,对活性组分造成覆盖,从而使表面活性Fe减少。因此,过多地加入铈不利于CO在铁表面上的配位吸附。
2.3 CO2-TPD研究
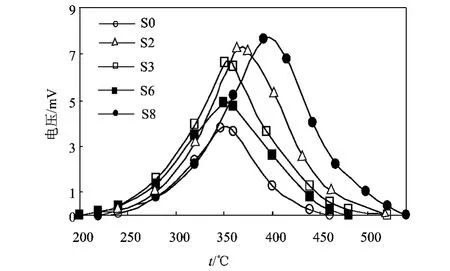
图5 各催化剂的CO2-TPD图
CO2作为表面碱度的探针分子,可以说明催化剂表面碱性强弱。图5是各催化剂的CO2-TPD图谱,可见随着CeO2含量的增加,脱附峰温及峰面积都有增加,也即CO2在催化剂表面上的吸附量增加。由于CeO2是强碱性的稀土氧化物,故随着CeO2含量的增加,催化剂表面碱性也在逐渐增强。CO2为一酸性气体,其在催化剂表面上的吸附与催化剂表面碱性有关,碱性增强利于CO2的吸附。但需要说明,对于变换反应来说,CO2是产物分子。如果生成的CO2在催化剂表面吸附过强,以致不能及时脱附,必然会影响反应进行。所以,催化剂中CeO2含量显然不易过大。
2.4 BET结果
表4给出了氮吸附法测得的比表面积。由表4可见,比表面积随着CeO2含量的增加有一最大值,对应于质量分数3%的CeO2,说明此种情况下CeO2对主剂分散度最大。如前所述,当催化剂中加入较多CeO2时,本身易集结,并对活性组分造成覆盖。这反而削弱了对主剂的分散,比表面积也随之降低。
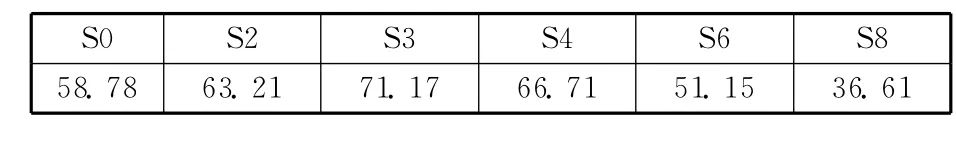
表4 催化剂样品的BET比表面积 m2/g
2.5 氧化铈的助催化作用分析
从以上研究结果看,少量氧化铈的加入明显改善了催化剂吸附CO性能与氧化性能。结合比表面积的表征结果,显然看出,氧化铈具有结构助剂的作用。众所周知,催化剂性能的改善与其比表面积有着密切的关系。如果氧化铈仅具有结构助剂的作用,则TPO的比脱氢量、TPD的比脱附量应与氧化铈含量有线性关系,但结果并不如此。图6至图8是CO-TPD的比脱附量、TPO的比脱氢量与CeO2含量的关系。另外,为了更好地对比说明,H2-TPR中比耗氢量与CeO2含量的关系[13]也在文中给出,见图9。
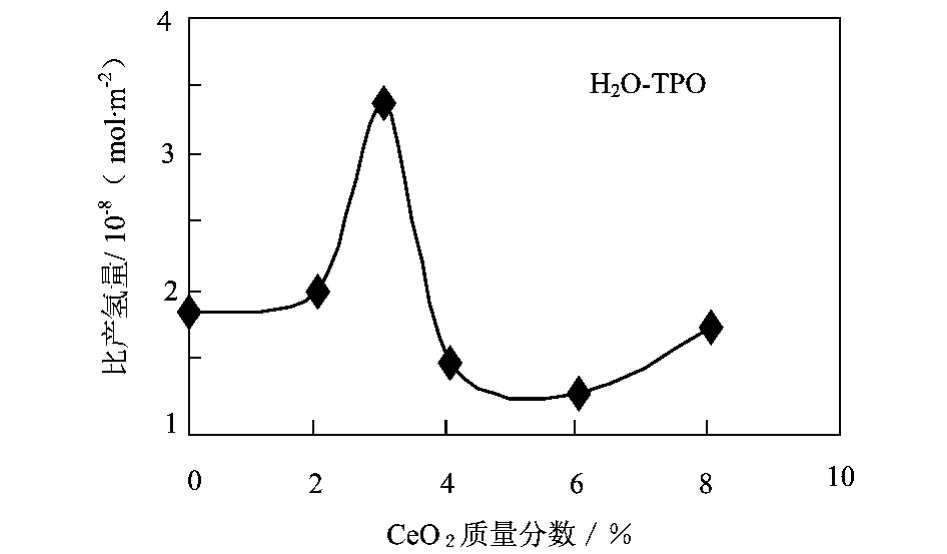
图6 H2O-TPO中比脱氢量与CeO2含量的关系
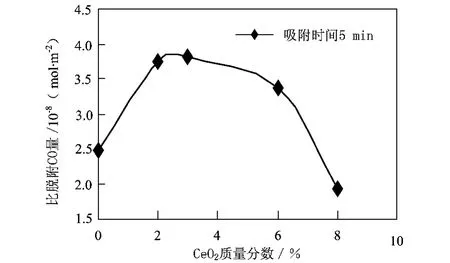
图7 CO-TPD中比脱CO量与CeO2含量的关系
可见,以上这些参数并不与CeO2含量成线性关系,而是呈一极值出现,这一极值对应于质量分数3%的CeO2含量。这又显示了CeO2电子型助催化作用。也就是说CeO2在中变催化剂中扮演着结构与电子型助剂的双重作用。
3 结论
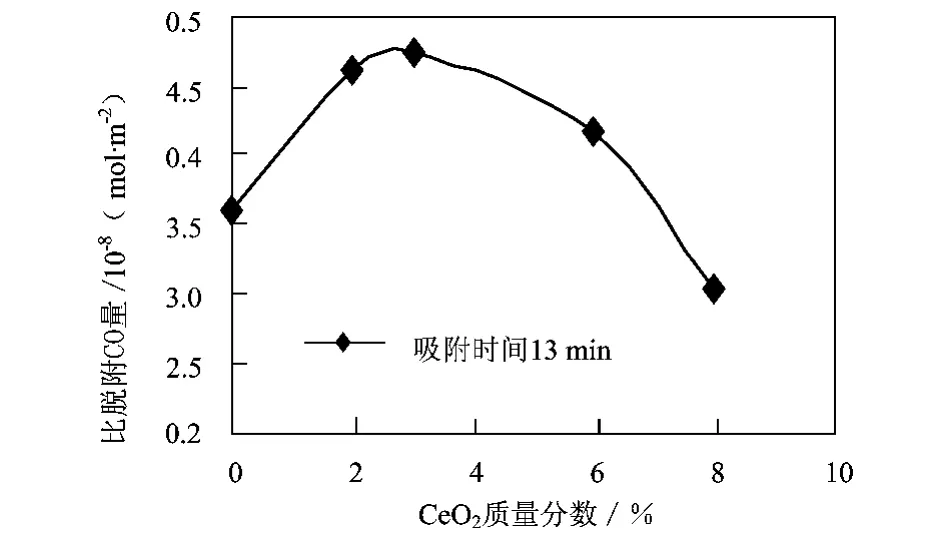
图8 CO-TPD中比脱CO量与CeO2含量的关系
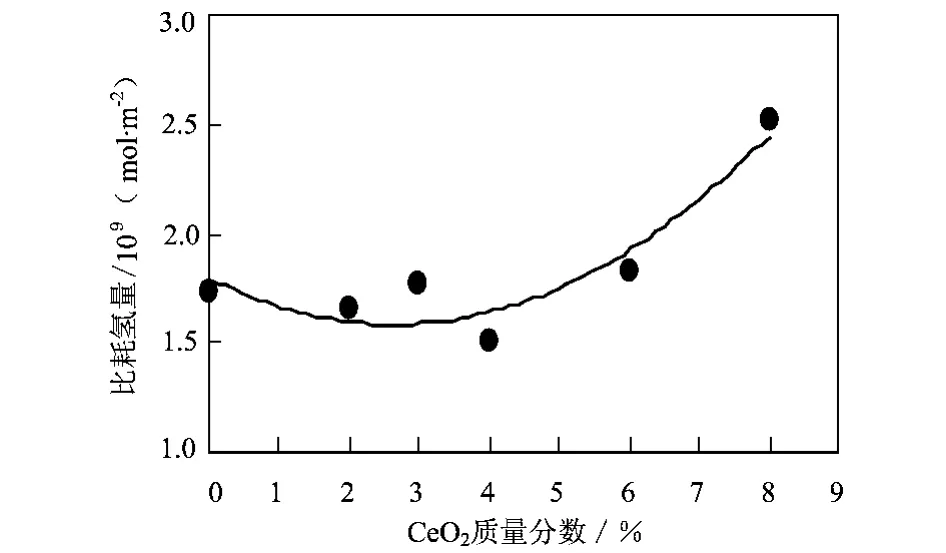
图9 H2-TPR中比耗氢量与CeO2含量的关系
1)在铁铬中变催化剂中加入氧化铈,可明显改变催化剂的吸附性能与氧化性能。当CeO2质量分数为3%时,对催化性质的改善作用最显著。
2)CeO2在铁铬变换催化剂中具有结构型与电子型助剂的双重作用。
3)少量CeO2的加入可以提高主剂分散度,增加催化剂的比表面积。但大量加入则会使CeO2本身积聚在催化剂表面,并对活性组分造成覆盖,反而降低了催化剂的比表面积。
[1] Fu Q,Kudriavtseva S,Saltsburg H,Flytzani-Stephanopoulos M.Gold-ceria catalysts for low-temperature water-gas shift reaction[J].Chemical Engineering Journal,2003,93:41-53.
[2] 董跃,赵钰琼,张永发.CO催化变换制氢反应机理及传统变换催化剂研究进展[J].山西节能与能源,2009(2):67-71.
[3] Rhodes C,Williams B P,King F,Hutchings G J.Promotion of Fe3O4/Cr2O3high temperature water gas shift catalyst[J].Catalysis Communications,2002(3):381-384.
[4] 郑起,徐建本,魏可镁.无铬CO高温变换催化剂的研究——氧化铬对高变催化剂结构与性能的影响[J].催化学报,1998,19(5):19-22.
[5] Doppler G,Ambach E.Physical and catalytic properties of high temperature water-gas shift catalysts based upon iron chromium oxides[J].Applied Catalysis,1988,40:119-130.
[6] Gorte R J,Zhao S.Studies of the water-gas-shift reaction with ceria-supported precious metals[J].Catalysis Today,2005,104:18-24.
[7] 关业军,李灿.氧化铈的氧化还原性能对VOx/CeO2催化剂催化氯苯氧化性能的影响[J].催化学报,2007,28(5):392-394.
[8] 欧晓佳,程极源.金属氧化物对低铬铁系催化剂结构和性能的影响[J].天然气化工,1998,23(6):12-15.
[9] 郑起,徐建本,魏可镁.无铬CO高温变换催化剂的研究——过渡元素对高变催化剂结构与性能的影响[J].催化学报,1999,20(1):21-24.
[10] 马红钦,张继炎,谭欣.铁基高温变换催化剂制备的穆斯堡尔谱研究[J].催化学报,1995,16(5):365-370.
[11] 胡延平,金恒芳.BX型中变催化剂的晶型研究[J].内蒙古大学学报,1993,12(1):59-65.
[12] 华金铭,郑起,林性贻,魏可镁.水煤气变换催化剂研究新进展[J].分子催化,2004,18(1):68-80.
[13] 李晓东,谈世韶,郭汉贤.含氧化铈铁铬系中变催化剂还原行为的研究[J].太原工业大学学报,1993,24(3):55-62.
[14] 李晓东,谈世韶,郭汉贤.氧化铈催化作用研究探讨[J].太原工业大学学报,1992,23(4):21-26.
[15] 梁斌,张鎏,王嘉福.助剂对铁基CO变换催化剂催化性能的影响[J].石油化工,1992,21(3):174-178.
猜你喜欢
小学生学习指导(高年级)(2022年3期)2022-03-29
烟台大学学报(自然科学与工程版)(2021年1期)2021-03-19
今日农业(2020年20期)2020-11-26
小学生学习指导(高年级)(2019年4期)2019-11-27
石油石化绿色低碳(2019年6期)2019-02-13
小学生导刊(高年级)(2017年2期)2017-06-10
浙江大学学报(工学版)(2016年11期)2016-06-05
Coco薇(2016年2期)2016-03-22
小学生导刊(高年级)(2016年1期)2016-01-29
中国资源综合利用(2016年4期)2016-01-22
