
再生障碍性贫血发病机制与治疗的探讨
2012-04-06 06:34刘文励房明浩
河南大学学报(医学版) 2012年3期
刘文励,房明浩
(华中科技大学同济医学院附属同济医院 血液内科,湖北 武汉 430030)
再生障碍性贫血发病机制与治疗的探讨
刘文励,房明浩
(华中科技大学同济医学院附属同济医院 血液内科,湖北 武汉 430030)
再生障碍性贫血是以全身血细胞减少为主要表现的造血功能衰竭性疾病。造血干细胞减少或缺损、T淋巴细胞功能异常亢进、细胞毒性T淋巴细胞直接杀伤和淋巴因子介导的造血干细胞过度凋亡引起的骨髓衰竭是该病的主要发病机制;造血微环境支持功能缺陷与再障的发生关系密切;流行病学资料显示,再障的发生也与特定的HLA相关。结合英国血液病学标准委员会公布的《再障诊断治疗指南》及中华医学会血液学分会红细胞疾病学组公布的《再障诊断治疗专家共识》,对再生障碍性贫血的诊断、治疗进行了分析和探讨,治疗中除了适当输入红细胞、血小板悬液及预防、治疗感染外,还应采取传统治疗以外的针对性治疗方案,以保证更好的疗效。
造血干细胞;T淋巴细胞;造血微环境;HLA;再障治疗方案
再生障碍性贫血(aplastic anemia,AA,简称再障)是多种病因引起的造血功能衰竭性疾病,红骨髓总容量减少,代之以脂肪髓,骨髓中无恶性细胞浸润,无网硬蛋白增生,临床上以全血细胞减少为主要表现的一组综合征。继发性再障可能的病因有:①药物:如各种抗肿瘤药物的毒性作用,与药物剂量有关,或是药物的特异性反应,与剂量无关,常见的有氯霉素、砷、金制剂等。②病毒感染:肝炎病毒、微小病毒B19等的感染。③辐射:长期接触X线,放射性核素等。④化学毒物:酚类,杀虫剂,农药,苯及其代谢产物均可抑制骨髓。⑤免疫因素:再障可继发于胸腺瘤、系统性红斑狼疮和类风湿性关节炎等,患者血清中可找到抑制造血干细胞的抗体。英国血液病学标准委员会(BCSH)[1-2]2008年公布了《再障诊断治疗指南》,中华医学会血液学分会红细胞疾病学组[3]公布了我国《再障诊断治疗专家共识》,要求我们临床各级医师应重视原发性获得性再障的规范化诊断和治疗,以争取更好的疗效。
1 再障的发病机制
1.1 造血干细胞减少或缺陷
许多再障患者用正常人造血干细胞成功地骨髓移植,显示出干细胞异常或缺陷是其发病的原因之一。再障患者骨髓CD34+细胞较正常人明显减少,骨髓细胞呈现造血不良表现,启动细胞(LTC-IC)明显减少或缺乏,CFU-GM、CFU-E形成能力较正常显著降低。
1.2 T淋巴细胞、细胞毒性T淋巴细胞和淋巴因子介导的造血干细胞异常是再障的主要发病机制[4-5]
1.2.1 特异性免疫紊乱 免疫抑制治疗临床疗效良好,如抗淋巴细胞球蛋白/抗胸腺细胞球蛋白(ALG/ATG)联合环孢霉素A(CSA)治疗再障,证实了本病发生的异常免疫损伤理论。介导异常免疫的T淋巴细胞分泌可溶性的造血负调控因子IFN-γ,激活Th1型细胞进一步分泌IFN-γ、IL-2、TNF-α等细胞因子。造血负调控因子通过诱导造血干细胞表面Fax表达增高,在促凋亡因子的协同作用下通过Fas/FasL途径导致造血干细胞凋亡。CD8+T细胞内IFN-γ水平的变化与免疫抑制治疗的疗效相关,是再障复发的可靠预测指标之一。
1.2.2 调节性T细胞缺陷 调节性T细胞(Tregs)是以细胞表面表达CD4和CD25,细胞内表达转录因子FOXP3为特征,通过抑制自身反应性T细胞而抑制自身免疫的发生和发展。转录因子NFAT1与FOXP3启动子结合后诱导其表达。再障患者均有Tregs的降低,FOXP3蛋白和mRNA水平也明显降低,NFAT1蛋白水平低至测不出[6]。CD4+CD25+Tregs细胞在诱导和维持自身免疫耐受性和阻止自身免疫中起着重要作用。Tregs能够抑制和调节CD4+和CD8+T细胞的活化和增殖,起到负调节作用。有研究发现,再障患者的Tregs细胞数量明显减少,Tregs细胞缺乏与自身免疫性骨髓衰竭明显有关。再障治疗后获缓解者,其Tregs的输注可改善淋巴细胞输注诱发的全血细胞减少。
1.2.3 T-bet表达增加 T-bet选择性地表达于Th1细胞,T-bet在再障中表达上调,T-bet蛋白与IFN-γ启动子区结合,是IFN-γ基因强有力的转录激活剂,诱导IFN-γ的产生。在Th1细胞的分化中起决定性作用。T-bet还能将分化中的效应性Th2和已完全分化的Th2细胞逆转为Th1,产生大量IFN-γ,抑制 Th2型细胞因子(如IL-4、IL-5等)的产生。
1.2.4 B淋巴细胞功能紊乱 再障主要与T细胞功能紊乱有关,但同样也发现了自身抗体。Hirano等发现39%的再障患者存在抗kinectin抗体,正常人及其他自身免疫性疾病中未检出该抗体,可能该抗体为再障所特有。Feng等发现抗地西泮结合相关蛋白 1(diazepam-binding inhibitor-related protein 1,DRS-1)抗体与再障免疫机制关联,携带DRS-1抗体的再障患者对免疫抑制剂治疗效果较好。约37%的再障患者可检测到抗膜突蛋白(Moesin)抗体,该抗体可影响造血细胞的功能和活力。有学者[7]认为,抗膜突蛋白抗体、PNH克隆和抗DRS-13种指标的联合检测对评估再障的免疫发病机制有帮助。
1.3 造血微环境支持功能缺陷
造血微环境包括基质细胞及其分泌的细胞因子,起支持造血细胞增殖及促进各种细胞生长发育的作用。已发现再障骨髓成纤维细胞集落形成单位(CFUF)和基质细胞产生的集落刺激活性(CSA)降低。中国医学科学院血液学研究所观察到再障骨髓基质细胞萎缩、脂肪化、静脉窦壁水肿、出血、毛细血管坏死、CFUF减少,急性再障较慢性再障损伤更严重。
1.4 遗传因素
流行病学资料发现,再障也与特定的HLA基因相关。再障患者常有HLA-DR2型抗原连锁倾向,儿童再障HLA-DPW3型抗原显著增高,患者家属中常有造血祖细胞增殖能力明显降低。
端粒位于线性染色体的末端,由5~15kb的重复序列(前导链TTAGGG,滞后链CCCTAA)组成,维持染色体的完整性。端粒长度的维持需要端粒酶,端粒酶主要由3种组分构成:端粒酶RNA组分(Telomerase RNA component,TERC)、逆转录酶组分(telomerase reverse transcriptase,TERT)、端粒酶相关蛋白(telomerase associated protein,TP)。约1/3获得性再障存在端粒DNA长度的缩短,并推测因端粒酶活性降低所致。约10%再障患者发现端粒酶基因突变,主要为TERC或TERT基因突变[8]。TERC基因突变主要集中于它的假结节区、CR4-CR5区,突变可能通过影响TERC与TERT分子之间的结合而降低端粒酶活性。TERT分子各结构域内均检测到再障发病相关突变基因[9],如位于逆转录酶区的突变Y772C(第772位半胱氨酸取代酪氨酸)、位于C端结构域的突变V1090m(蛋氨酸取代缬氨酸)等。如1例26岁男性再障患者,发现TERT分子N端结构域突变K570N(天冬酰胺取代赖氨酸),其外周血粒细胞端粒DNA长度3.8kb(同龄正常人群8.6kb),淋巴细胞端粒DNA长度3.1kb(正常人群7.5kb),体外转染K570N突变的重组细胞端粒酶活性明显降低仅为野生型细胞的1%。TERT突变基因携带者体内造血细胞数量较没有基因突变者显著减少。端粒重复结合因子1(telomeric tepeat binding factors 1,TRF1)与端粒DNA结合,抑制端粒与端粒酶结合时端粒酶末端弯曲成襻,Savage等[10]发现端粒重复结合因子1第36912位核苷酸胸腺嘧啶取代胞嘧啶所引起的突变可能是再障发病的危险因素。在免疫抑制剂治疗中观察到,端粒较短者再障复发的可能性更高,发生急性髓细胞性白血病(acute myelo-cytic leukemia,AML)的风险增加,骨髓细胞染色体不稳定性增加。
2 再障的实验室检查
初诊再障患者需要进行的实验室检测指标有:①全血细胞计数、网织红细胞计数、血涂片检查。②胎儿血红蛋白(HbF)测定。③骨髓穿刺(髂骨和胸骨细胞学,中性粒细胞碱性磷酸酶,糖原染色,铁染色及小巨核细胞酶标)及活检(骨髓病理和网状纤维染色),造血祖细胞培养(CFU-E、BFU-E、CFU-GM、CFUF)是必须的检查。④肝脏功能及病原学检查。⑤血叶酸、VitB12水平、SI、TIBC、UIBC、ISAT+铁蛋白(SF)、血清转铁蛋白、可溶性转铁蛋白受体检测。⑥外周血流式细胞免疫分型(T细胞亚群和大颗粒淋巴细胞检测)以及检测CD55、CD59,了解是否有PNH克隆,进行尿含铁血黄素检测有无血管内溶血。⑦自身抗体筛选(抗核抗体、抗dsDNA抗体)及免疫全套,细胞因子(IL-2、IL-4、TNF、INF-γ)水平,红细胞生成素(EPO)水平检测。⑧胸部CT或X片可以排除肺部感染,并且可留着作对照。⑨腹部超声,了解肝、脾、淋巴结有无肿大,较为年轻的患者肾脏异常提示可能为Fanconi’s贫血。⑩常规细胞遗传学检查及荧光原位杂交(fluorescen in situ hybridization,FISH),特别注意5号、7号染色体有无异常。进行遗传性疾病的筛选,检测外周血淋巴细胞是否存在染色体断裂,以排除Fanconi’s贫血。对于具备先天性角化不良症状以及对免疫治疗不敏感者进行外周血端粒酶DNA长度及融合基因检测(DKC1,TERC,TERT)。
3 再障的诊断标准
3.1 一般标准
①全血细胞减少,网织红细胞绝对值减少。②一般无肝脾肿大。③骨髓至少一个部位增生减低或重度减低(如增生活跃,须有巨核细胞明显减少),骨髓小粒非造血细胞增多,骨髓活检提示造血组织减少,脂肪组织增加。④排除引起全血细胞减少的其他疾病。⑤抗贫血药物治疗无效。
3.2 重型再障(SAA)的诊断标准
①临床表现:发病急,贫血进行性加剧,常伴随严重感染、内脏出血。②血象:除血红蛋白下降较快外,须具备下列3项:网织红细胞<1%,绝对值<15×109/L;白细胞明显减少,中性粒细胞绝对值<0.5×109/L;血小板<20×109/L。③骨髓象:多部位增生减低,三系造血细胞明显减少,非造血细胞增多,如增生活跃,有淋巴细胞增多;骨髓小粒中非造血细胞及脂肪细胞增多。
3.3 非重型再障(NSAA)的诊断标准
发病缓慢,以贫血表现为主,感染、出血较轻;血象和骨髓象未达到重型再障的诊断标准。
4 鉴别诊断
4.1 溶血性疾病
最主要的是阵发性睡眠性血红蛋白尿症(paroxysmal nocturnal hemoglobinuria,PNH),典型PNH有血红蛋白尿发作,易鉴别。不典型者无血红蛋白尿发作,全血细胞减少,骨髓可增生减低,易误诊为再障,但该病主要特点是,动态随访,终能发现PNH造血克隆。对于受累红细胞<10%的PNH,溶血检查常为阴性,不能检测出PNH克隆的存在。通过流式细胞术检测造血细胞GPI锚链蛋白(CD55、CD59)的表达水平是诊断PNH的敏感方法。目前认为,PNH克隆是从粒细胞逐渐发展到红细胞,首先受累的是造血祖细胞,当外周血细胞尚无GPI锚链蛋白分子缺陷时,骨髓细胞可能已有GPI锚链蛋白分子缺陷,因此,检测骨髓细胞比外周血细胞更有意义。部分再障患者也会出现少量PNH克隆,其表达水平可以保持不变、减少、消失或是增加。若这些患者有实验室或临床证据表明存在溶血,应诊断为PNH。尿含铁血黄素试验阳性,提示存在长期血管内溶血,有利于PNH的诊断。网织红细胞计数、间接胆红素水平、转氨酶和乳酸脱氢酶定量对于评价PNH的溶血也有一定作用。
Evans综合征和免疫相关性全血细胞减少症的鉴别。前者可检测外周血细胞自身抗体(coombs试验阳性),后者可检测骨髓未成熟血细胞膜上自身抗体。这两类血细胞减少患者Th2细胞比例增高,CD5+的B淋巴细胞比例增高,血清IL-4水平增高,对肾上腺糖皮质激素和/或大剂量静脉免疫球蛋白治疗反应好。
4.2 免疫系统疾病
B细胞功能亢进的疾病,如系统性红斑狼疮、免疫相关性血细胞减少症,可以产生抗造血细胞的自身抗体,引发造血功能衰竭。系统性红斑狼疮还可引起骨髓纤维化,疑为系统性红斑狼疮等结缔组织病应检查抗核抗体及抗dsDNA抗体等。
4.3 低增生性MDS
低增生性骨髓增生异常综合征 (myelodysplastic syndromes,MDS)很难与再障相鉴别,但低增生性MDS周围血单核细胞往往增多,并可见幼稚细胞;骨髓两系或三系细胞呈病态造血,部分患者骨髓活检显示网硬蛋白增生及不成熟前体细胞异常定位(ALIP)现象。另外,通过有核红细胞糖原染色、小巨核酶标、白血病集落形成单位(CFU-L)及染色体核型细胞遗传学检查等亦有助于二者的鉴别。因骨髓增生低下,细胞数少,难以获得足够的中期分裂相细胞,FISH方法可提高检出率。在儿童再障中出现遗传学异常,尤其是+7常提示为MDS。在疾病的过程中可能会出现异常细胞遗传学克隆。目前推荐的FISH 套餐是5q31、CEP7、7q31、CEP8、20q、CEPY和p53。2008年WHO关于MDS诊断分型标准中认为,单有-Y,-8或20q-的难治性血细胞减少者,若无明确病态造血,不能依遗传学异常而诊断为MDS,应动态观察。对此的解释是,这些患者常常对免疫抑制治疗有较好效果。
4.4 低增生性ALL
低增生性急性淋巴细胞白血病(acute lymphoblastic leukemia,ALL)发病率占儿童ALL的1%~2%。有些患儿可能在骨髓衰竭后3~9个月进展为ALL,中性粒细胞减少较血小板减少更为严重。白细胞减少的低增生性ALL可呈慢性过程,早期肝、脾、淋巴结未肿大,外周血全血细胞减少,骨髓增生减低。仔细观察血象及多部位骨髓象,可发现原始淋巴细胞明显增多,骨髓活检和免疫分型及TCR、IgH基因检测有助于与再障的鉴别诊断。
4.5 低增生性AML
白细胞减少的白血病和低增生性白血病,早期肝、脾、淋巴结不肿大,外周全血细胞减少,易与再障混淆。仔细观察血象及多部位骨髓,可发现原始粒细胞或原始(幼)单核细胞明显增多。部分急性早幼粒细胞白血病,伴t(8;21)易位的NALL(M2)可有全血细胞减少,骨髓分类和流式细胞MPO阳性等有助于鉴别。
4.6 毛细胞性白血病
毛细胞性白血病表现为全血细胞减少,伴有持续性的单核细胞减少,骨髓穿刺术可能出现“干抽”现象。骨髓活检可以见到毛细胞浸润以及网硬蛋白增加。免 疫 表 型 显 示 CD20+、CD22+、CD11C+、CD25+、FMC7+、CD103+、CD5-、CD10- 和 CD23-肿瘤细胞。30%~40%患者可能出现脾脏肿大。
4.7 肿瘤骨髓转移
晚期肿瘤(胃癌、肺癌及卵巢癌)发生骨髓转移浸润,可导致造血功能降低,血象表现为全血细胞减少。骨髓穿刺和活检检查可见到转移的肿瘤细胞。部分患者可显示原发病的症状与体征,通过免疫分型、基因重排将有助于鉴别诊断。
4.8 脾脏功能亢进症
脾脏功能亢进症所致的血细胞过度消耗,如肝硬化、结缔组织病、恶性淋巴瘤等均表现为全血细胞减少,易与再障混淆。这类疾病脾脏均明显肿大,骨髓检查显示骨髓造血细胞增生活跃,并可发现相应的异常细胞。
4.9 骨髓纤维化
慢性病例常有脾脏肿大,表现为全血细胞减少和骨髓增生减低,骨髓常干抽。骨髓活检见到网硬蛋白增加和纤维细胞。骨髓纤维化因出现髓外造血,血涂片可以见到不成熟造血细胞。无脾脏肿大的骨髓纤维化继发于恶性肿瘤的可能性大。
4.10 先天性再障
范科尼贫血(FA)常称为先天性再障,是一种遗传性干细胞质异常性疾病。表现为一系、两系或全血细胞减少,可伴发育异常(皮肤色素沉着、骨骼畸形、器官发育不全等),高风险发展为MDS、ALL及其他各类肿瘤性疾病。实验室检查可发现“范可尼基因”、外周血细胞染色体受丝裂酶素C或DBA试剂作用后极易断裂[11-12]。因有较大年龄的范科尼贫血病例报道,其筛查的上限年龄可达50岁。先天性角化不良可以通过典型临床特征和基因突变加以鉴别。
4.11 感染
分支杆菌,尤其是非典型分支杆菌感染会出现全血细胞减少和骨髓增生低下。骨髓检查还可发现肉芽肿、纤维化、骨髓坏死等。嗜酸性坏死常见于非典型结核杆菌感染。疑为结核者,应送骨髓液行分支杆菌培养。
5 再障的治疗
除根据病情适当输注红细胞、血小板悬液(应输注经辐照或白细胞滤过的制剂),预防及治疗感染外,对再障的治疗,还应采取针对性的治疗方案。
5.1 输血依赖性的非重型再障(NSAA)
患者应尽早接受标准的免疫抑制剂治疗(immunosuppressive therapy,IST),经过3~6个月治疗有效果的患者,维持CSA治疗>6个月或外周血细胞水平完全恢复后CSA缓慢减量;如经过4~6个月抗胸腺球蛋白(antithymocyte globulin,ATG)或抗淋巴细胞球蛋白(antilymphocyte globulin,ALG)+环孢素A(cyclosporin A,CSA)治疗无效果者,年龄在50岁以下可考虑骨髓移植,或者考虑行第二疗程ATG治疗,如第二疗程ATG治疗4~6个月仍无效或疾病进展为重型再生障碍性贫血(severe aplastic anemia,SAA),则按SAA治疗。
端粒DNA缩短或端粒酶突变的再障患者,对雄激素治疗有一过性的反应,雄激素通过自身芳香化为雌激素及雌激素的受体途径,激活造血干细胞的端粒酶活性,故肝功能好者可加用雄激素安特尔治疗,口服40mg,2~3次/d。
5.2 重型再障治疗策略
重型再障宜及早行HLA相合同胞供体的allo-BMT或ATG+CSA的强化IST:①<40岁患者,选择HLA相合同胞供体的allo-BMT;未找到合适供体的行免疫抑制治疗(ATG+CSA)。②>40岁患者,选择ATG十CSA治疗。③接受ATG十CSA治疗,经过4个月治疗有效果的患者,维持CSA治疗>6个月或外周血细胞水平完全恢复后CSA缓慢减量,如经过4个月(ATG+CSA)治疗无效果者,可进行第2疗程的(ATG+CSA)治疗,或者考虑无关供体配型骨髓移植,见图1。
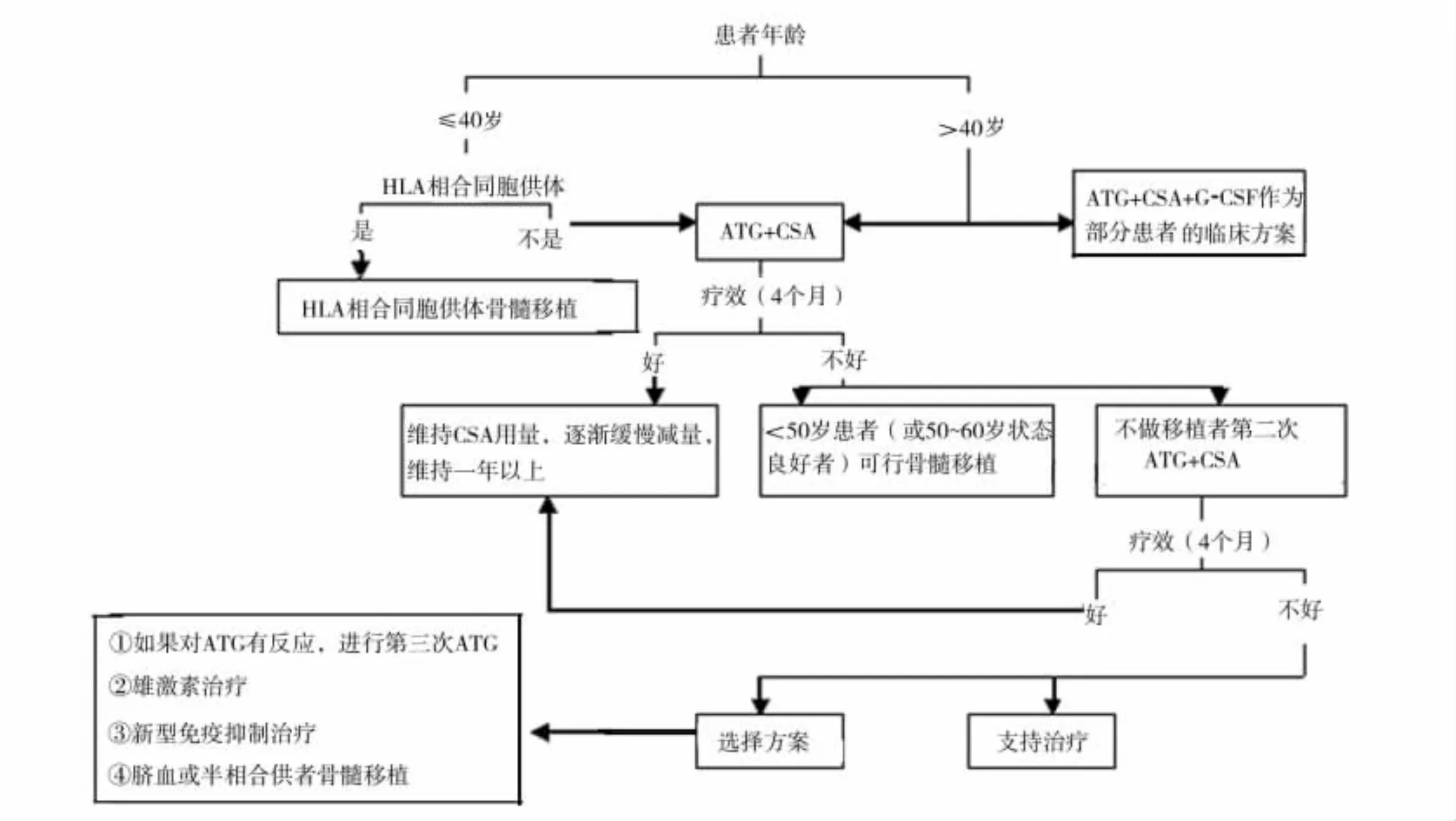
图1 重型再障的治疗流程
5.3 SAA的同胞供者异基因骨髓造血干细胞移植
适应症:①重型再障患者年龄小于40岁,最大年龄不应超过45岁。②有HLA相合的同胞兄弟姐妹做供体。③既往无或少许输注血液制品史的早期患者。④无明显感染迹象。
年龄<30岁的年轻患者的预处理:采用非清髓、高强度免疫抑制方案以预防移植排斥和移植物抗宿主病(graft versue host disease,GVHD)。40岁以上的患者如果进行骨髓移植,应给于低强度的预处理。在再障亲缘全相合造血干细胞移植(HSCT)中不建议进行照射预处理。
5.4 再障的免疫抑制治疗[13-14]
适应症:①是依赖输血的非重型再障患者的一线治疗。②不依赖输血的非重型再障患者,有明显的粒细胞缺乏伴随继发感染的高风险。③年龄>40岁的重型再障患者。④<40岁年龄缺乏HLA相合同胞供者的重型再障患者。IST疗效反应率似不受病因学(如肝炎、病毒接触史、PNH/AA综合征)的影响,但单用ATG治疗SAA反应率明显低于ATG+CSA;ATG+CSA治疗NSAA反应率明显高于单用CSA者。由于联合治疗的疗效优于任何单一用药,ATG+CSA的联合方案已成为目前再障的标准疗法。具体用法为马 ATG 20mg·kg-1·d-1,连用4d或兔 ATG 3.5mg·kg-1·d-1,连用5d,联合CSA 5mg·kg-1·d-1,分2次口服,连续6个月。5年总体生存率(overall survival,OS)75%~80%。ATG治疗反应一般发生于6个月内,通常在l~2个月可观察到病情的好转,2~3个月脱离血制品输注,但也有较晚起效者。ATG治疗3个月有效率50%,治疗6个月有效率70%~75%。IST有效,也说明这些患者发病可能源于自身免疫。IST有效者应持续服用CSA,逐渐减量至维持剂量,早期或骤然停用CSA可致病情加重或反复。当CSA用至6个月撤掉时,30%~35%的患者会复发,若延长应用CSA,并缓慢逐渐减量,复发的危险性约13%~16%。大约1/3的再障患者依赖CSA,需要小剂量长期维持。当第一疗程ATG治疗后复发,或者第一疗程没有反应,可给第二疗程ATG治疗,开始是马ATG,第二疗程则应改为兔ATG,复发患者疗效可达65%,第一疗程无效者第二疗程反应率约30%。
ATG常见近期不良反应包括急性过敏反应(发热、寒战、多形性皮疹、高血压、低血压等)、血清病反应(皮疹、非感染性发热、关节疼痛、肌肉痛、浆膜炎、淋巴结病等)。前者多发生于治疗最初的几天,后者则常发生于接受ATG输注后的14d内。防治以小剂量皮质类固醇激素为主。其他不良反应还包括引起血小板和中性粒细胞减少、肝肾功能损害、心律失常等。中性粒细胞减少可发生致命的感染。SAA患者接受强化IST缓解后,数年可能并发克隆性疾病,如PNH、MDS、AML及实体肿瘤等。IST治疗11年后,其发生率分别为PNH 10%,MDS或AML为8%,实体肿瘤为11%。7号染色体和8号染色体改变多见。对可能演变为MDS或AML的危险因素是:①重复的应用ATG。②年龄较大者。③在用ATG和CSA的同时长期用较大量的G-CSF。④短的端粒DNA长度。
5.5 免疫抑制治疗的疗效预测
IST无效的可能原因有:①IST治疗的剂量和疗程不充足,不标准。②不可逆的干细胞损伤。③非免疫介导的再障。
预测IST(ATG/CSA)疗效反应是目前SAA临床研究的热点领域之一,因为这可为IST进一步治疗(解救或替代治疗)方案的制订提供更多的信息,从而减少治疗的被动性和盲目性。①CSA血药浓度:起始治疗2周时CSA血药浓度与疗效反应相关。②IFN-γ水平:采用流式细胞术测定T淋巴细胞内IFN-γ的水平能区分出大多数治疗有效和无效的患者,IFN-γ的表达水平与临床进程密切相关。③HLA-DRI5表达和IST临床反应显著正相关。④伴有PNH克隆的SAA患者对IST治疗反应率较高,年轻且有HLA相合同胞供者的PNH-SAA患者IST有效率低,应首选移植,而PNH+SAA患者则宜首选IST。⑤极重型再障(VSAA)及rHuG-CSF治疗无反应者IST疗效欠佳,因此,宜首选HLA相合的同胞供者allo-BMT。⑥端粒DNA长度短的再障患者,IST初治也有效,但易复发,且易发生细胞遗传学异常,是演变为MDS或AML的危险因素。
[1]Marsh J C,Ball S E,Cavenagh J,et al.British Committee for Standards in haematology.Guidelines for the diagnosis and management of aplastic anaemia[J].Br J Haematol,2009,147(1):43-70.
[2]贺冠强,吴学琼,刘文励,等.再生障碍性贫血的诊断和治疗指南 [J].国际输血及血液学杂志,2009,32(4):356-365.
[3]中华医学会血液学分会红细胞疾病(贫血)学组.再生障碍性贫血诊断治疗专家共识 [J].中华血液学杂志,2010,31(11):790-792.
[4]Young N S,Scheinberg P,Calado R T.Aplastic anemia[J].Curr Opin Hematol,2008,15(3):162-168.
[5]Young N S,Bacigalupo A,Marsh J C.Aplastic anemia:pathophysiology and treatment[J].Biol Bolld Marrow Transplant,2010(16):110-125.
[6]Solomou E E,Rezvani K,Mielke S,et al.Deficient CD4+CD25+FOXP3+T regulatory cells in acquired aplastic anemia[J].Blood,2007,110(5):1603-1606.
[7]Takamatsu H,Espinoza J L,Lu X,et al.Anti-moesin antibodies in the serum of patients with aplastic anemia stimulate peripheral blood mononuclear cells to secrate TNF-alpha and IFN-gamma J[J].Immunol,2009,182(1):703-710.
[8]Yamaguchi H,Calado R T,Ly H,et al.Mutations in TERT,the gene for telomerase reverse transcriptase,in aplastic anemia [J].N Engl J Med,2005,352(14):1413-1424.
[9]Calado R T.Telomeres and marrow failure[J].Hematology Am Soc Hematol Educ Program,2009:338-343.
[10]Savage S A,Calado R T,Xin Z T,et al.Genetic Variation in telomeric repeat binding factors 1and 2in aplastic anemia[J].Exp Hematal,2006,34(5):664-671.
[11]Dokal L,Vulliamy T.Inherited aplastic anemias/bone marrow failure syndromes[J].Blood Rev,2008,22(3):141-153.
[12]Shimamura A,Alter B P.Pathophysiology and management of inherited bone marrow failure syndromes [J].Blood Rev,2010,24(3):101-12.
[13]Brodsky R A,Chen A R,Dorr D,et al.High-dose cyclophosphamide for severe aplastic anemia:long-term follow-up[J].Blood,2010,115(11):2136-2141.
[14]Risitano A M.Immunosuppressive therapies in the management of immune-mediated marrow failures in adults:where we stand and where we are going[J].Br J Haematol,2011,152(2):127-140.
[责任编辑 时 红]
R566.5
A
1672-7606(2012)03-0157-06
2012-06-12
猜你喜欢
西南医科大学学报(2023年1期)2023-04-06
河南科学(2020年3期)2020-06-02
云南医药(2019年3期)2019-07-25
癌变·畸变·突变(2019年3期)2019-06-03
铜仁学院学报(2018年6期)2018-07-05
分析化学(2017年12期)2017-12-25
恋爱婚姻家庭·养生版(2016年9期)2016-09-07
海南医学(2016年8期)2016-06-08
恋爱婚姻家庭·养生版(2016年5期)2016-05-06
中国继续医学教育(2015年3期)2016-01-06
