
以D-果糖为原料利用新型异构酶转化生产D-阿洛糖
2012-02-26 13:20:52柏玮朱玥明门燕李晓波何森健孙媛霞
生物工程学报 2012年4期
柏玮,朱玥明,门燕,李晓波,何森健,孙媛霞
天津工业生物技术研究所 工业酶国家工程实验室,天津 300308
在碳水化合物研究领域中,稀少糖是一类重要的研究内容。国际糖协会 (ISRS) 对稀少糖(Rare sugar) 的定义为“在自然界中存在但含量极少的一类单糖及其衍生物”[1]。这类糖大多可作为一种低热量、低吸收的甜味剂,并具有独特的生理学功能,在膳食、保健、医药等领域中发挥着非常重要的功能。例如,D-阿洛酮糖可以改善肠道菌群、降低血糖、抗龋齿等[2-4],D-阿洛糖具有抑制活性氧、抑制癌细胞增殖等功能等[5]。
日本的研究学者Izumori[6]于2002年建立了稀少糖的生物转化生产策略,即Izumoring方法。该方法中利用酮糖差相异构酶 (Ketose epimerase)、醛糖异构酶 (Aldose isomerases) 和多元醇脱氢酶 (Poly dehydrogenase) 进行所有单糖及糖醇之间的相互转化。该策略提示给我们一种利用生物转化生产稀少糖的方法,即利用天然植物成分或食品工业中加工副产物淀粉、乳糖为原料经过现有成熟工艺获得木糖、果糖、半乳糖,再通过各种酶的作用获得稀少糖。在 Izumoring策略中,以天然原料为切入点,有一条关键的切入途径。即:淀粉→果糖→D-阿洛酮糖。
D-阿洛酮糖是 D-果糖的 C-3差相异构体。生物催化转化D-果糖生产D-阿洛酮糖,可以利用的酶包括:D-塔格糖-3-差相异构酶(D-tagatose 3-epimerase,缩写为 DTE) 和D-阿洛酮糖-3-差相异构酶 (D-psicose 3-epimerase,缩写为DPE)[7-9]。其中,DTE的最适底物是D-塔格糖,而DPE则在底物是D-阿洛酮糖时发挥最大的催化活力。目前为止被发现并表征的D-阿洛酮糖生产酶只有两种DTE[7,15]和两种DPE[8,14]。通过比对发现,来源于C. cellulolyticum H10的DPE基因与已经发现的根癌农杆菌 Agrobacterium tumefaciens的DPE基因具有60%的相似性。因此,C. cellulolyticum DPE有潜力成为生产D-阿洛酮糖的第5种C-3-差相异构酶。
D-阿洛酮糖与 D-阿洛糖之间的相互转化,是一个醛酮异构反应。1998年,Bhuivan等首次利用基因工程获得的假单胞菌 Pseudomonas stutzeri的 L-鼠李糖异构酶 (L-rhamnose isomerase,缩写为 L-RhI) 进行酶固定化和循环转化生产 D-阿洛糖。将 L-RhI固定在壳聚糖BCW-2603珠载体上,加入20% D-阿洛酮糖溶液在40 ℃条件下进行反应,最终约40%转化为D-阿洛糖[10]。然而,L-RhI的最适底物并不是D-阿洛酮糖,而是 L-鼠李糖。大部分的 L-RhI因此并不具备催化生产 D-阿洛糖的能力,比如Escherichia coli L-RhI[11]、苍白芽胞杆菌Bacillus halodurans L-RhI[12]以及海栖热袍菌Thermotoga maritime L-RhI[13]。开发新的L-RhI,并研究不同来源的L-RhI催化生产D-阿洛糖的机理,因此变成一个十分有趣的课题。
本文克隆了来源于C. cellulolyticum H10的DPE基因以及来源于 Bacillus subtilis 168的L-RhI基因,并分别使其在宿主菌 B. subtilis及E. coli BL21 (DE3) 中得到了表达。接着,对表达后的DPE及L-RhI进行了纯化。最后对两种重组蛋白的催化转化能力进行了试验。打通了以D-果糖为原料转化生产 D-阿洛酮糖,最后得到D-阿洛糖的工艺路径。
1 材料与方法
1.1 材料
1.1.1 生化试剂
D-果糖、D-阿洛酮糖、D-塔格糖、D-山梨糖、D-阿洛糖等稀少糖均为分析纯试剂,购自Sigma Aldrich。蛋白胨、酵母粉、琼脂及其他试剂均为分析纯。
1.1.2 菌种与质粒
对于DPE基因的克隆来说,质粒与宿主菌分别为pMA5 (2) 及B. subtilis WB600。
对于L-RhI基因的克隆来说,质粒与宿主菌分别为pET-21a (+) 与E. coli BL21 (DE3)。
B. subtilis及 E. coli的培养基都是 Luria–Bertani (LB) 培养基。
1.2 方法
1.2.1 DPE基因在B. subtilis中的克隆与表达
根据C. cellulolyticum H10的DPE基因序列设计引物:上游引物pDPEF和下游引物pDPER。上下游引5¢端分别引入Nde Ⅰ和BamHⅠ的酶切位点 (表1)。并在C端插入了一段6×His标签,以简化其纯化步骤。
DPE的PCR基因片段和表达载体pMA5用限制性内切酶NdeⅠ和BamHⅠ进行酶切反应。回收后的目的片段与载体用 T4 DNA连接酶连接,连接产物pMA-DPE转化至B. subtilis WB600中进行克隆,PCR筛选阳性克隆,并用双酶切鉴定,挑取阳性转化子。
插入 pMA-DPE 的重组菌接入 LB-kanamycin培养基 (kanamycin终浓度50 mg/L)中,37 ℃、200 r/min培养16 h。
1.2.2 重组DPE蛋白的纯化
来源于C. cellulolyticum H10的DPE基因通过基因重组在B. subtilis WB600中表达,经过3个步骤对表达产物进行分离纯化,最终达到了电泳纯:
1) 粗提取:离心收集重组菌10 min (4 ℃,6 000×g),弃去上清,并用ddH2O洗涤菌体2次,离心20 min (4 ℃,12 000 r/min),弃去上清,用破碎缓冲液 (25 mmol/L Tris-HCl,300 mmol/L NaCl,40 mmol/L咪唑,pH 8.0) 悬浮,并在冰上超声破碎细胞,离心20 min (12 000 r/min,4 ℃)。

表1 PCR引物列表Table 1 PCR primers used in this study
2) 镍离子亲和层析:将上步所得离心上清经0.45 μm的膜过滤后,上样His-Trap镍离子螯合柱(平衡缓冲液:25 mmol/L Tris-HCl,300 mmol/L NaCl,40 mmol/L 咪唑,pH 8.0),以高咪唑含量的缓冲液进行线性洗脱 (25 mmol/L Tris-HCl,300 mmol/L NaCl,250 mmol/L 咪唑,pH 8.0)。收集目的蛋白。
3) 离子交换色谱:将上步所得溶液经超滤脱盐后,上样 Source 15Q阴离子交换柱 (GE Healthcare Biosciences) (平衡缓冲液:20 mmol/L Tris-HCl,pH 6.5),以高NaCl浓度的缓冲液进行线性洗脱 (20 mmol/L Tris-HCl,500 mmol/L NaCl,pH 6.5)。收集目的蛋白洗脱峰。
最后用20 mmol/L HEPES (pH 8.0) 的缓冲液将酶溶液超滤脱盐,并低温保存。
1.2.3 L-RhI基因在E. coli BL21中的克隆与表达
根据B. subtilis 168的L-RhI基因序列设计引物:上游引物pRHIF 和下游引物pRHIR。上下游引物5¢端分别引入BamHⅠ和XhoⅠ的酶切位点。并在C端插入了一段6×His标签,以简化其纯化步骤。
L-RhI的PCR基因片段和表达载体pET-21a (+) 用限制性内切酶BamHⅠ和 XhoⅠ进行酶切反应。回收后的目的片段与载体用T4 DNA连接酶连接,连接产物pET-21a-L-RhI转化至E. coli DH5α感受态中进行克隆,PCR筛选阳性克隆,并用双酶切鉴定,挑取阳性转化子转入E. coli BL21进行诱导表达。
插入 pET-21a-L-RhI的重组菌接入 LB-ampicillin培养基 (ampicillin终浓度 100 mg/L)中,37 ℃、200 r/min培养16 h。
1.2.4 重组L-RhI蛋白的纯化
经过3个步骤对重组L-RhI表达产物进行分离纯化,最终达到了电泳纯:
1) 粗提取:离心收集重组菌10 min (4 ℃,6 000×g),弃去上清,并用ddH2O洗涤菌体2次,离心20 min (4 ℃,12 000 r/min),弃去上清,用破碎缓冲液 (20 mmol/L Tris-HCl,1 mol/L NaCl,0.5%甘油,pH 8.0) 悬浮,并在冰上超声破碎细胞,离心20 min (12 000 r/min,4 ℃)。
2) 镍离子亲和层析:将上步所得离心上清经0.45 μm的膜过滤后,上样 His-Trap镍离子螯合柱 (平衡缓冲液:20 mmol/L Tris-HCl,1 mol/L NaCl,0.5%甘油,pH 8.0),以高咪唑含量的缓冲液进行线性洗脱 (20 mmol/L Tris-HCl,1 mol/L NaCl,0.5%甘油,200 mmol/L咪唑pH 8.0)。收集目的蛋白。
3) 离子交换色谱:将上步所得溶液经超滤脱盐后,上样DEAE Sepharose Fast Flow阴离子交换柱 (GE Healthcare Biosciences) (平衡缓冲液:20 mmol/L Tris-HCl,pH 6.0),以高NaCl浓度的缓冲液进行线性洗脱 (20 mmol/L Tris-HCl,500 mmol/L NaCl,pH 6.0)。收集目的蛋白洗脱峰。
最后用50 mmol/L Glycine-NaOH (pH 9.0)的缓冲液将酶溶液超滤脱盐,并低温保存。
本文所有纯化步骤所用系统均为 ÄKTA PurifierTM 100 (GE Healthcare Biosciences)。
1.2.5 DPE催化D-果糖转化为D-阿洛酮糖
底物溶液的配制:在 20 mmol/L HEPE (pH 8.0) 的缓冲液中,加入 D-果糖,配成终浓度10 g/L的溶液。
在500 μL的底物溶液中加入2.5 μg纯化后的DPE,于50 ℃的金属浴中反应。在100 ℃的沸水浴中加热5 min终止反应。
1.2.6 L-RhI催化D-阿洛酮糖转化为D-阿洛糖
底物溶液的配制:在50 mmol/L Glycine- NaOH (pH 9.0) 的缓冲液中,加入1 mmol/L MnCl2,再加入D-阿洛酮糖,配成终浓度10 g/L的溶液。
在1 mL的底物溶液中加入49.0 μg纯化后的L-RhI酶液,于 60 ℃的金属浴中反应。终止反应时加入100 μL三氯乙酸 (10%)。
1.2.7 糖的检测
仪器为安捷伦高效液相色谱仪 1200,分析柱:安捷伦 Zorbax 糖分析柱,流动相:75%乙腈 (以D-果糖为底物生成D-阿洛酮糖) 或78%乙腈 (以D-阿洛酮糖为底物生成D-阿洛糖),流速:1 mL/min,柱温:30 ℃,检测器:示差折光检测器。以Sigma公司生产的D-果糖、D-阿洛酮糖和 D-阿洛糖纯品为标准品,将上例得到的样品进行分析,上样量为5 μL。
2 结果与分析
2.1 DPE基因的表达与纯化
已有研究报道了来源于 C. cellulolyticum H10的DPE基因在E. coli中的表达[11]。然而结果表明,DPE在E. coli中的表达量有限,并且表达产物大部分是包涵体,不能直接用于糖反应。这对DPE基因的表达体系提出了一个新的挑战。用枯草芽胞杆菌作宿主菌,有以下几个优点:1) 枯草芽胞杆菌表达体系不需要诱导物的加入,一方面节约了成本,另一方面免去某些诱导物对人体的毒害;2) 利用枯草芽胞杆菌,可以构建分泌型的表达系统,将目的蛋白分泌到细胞外部,从而不再需要细胞的破碎步骤,简化了操作流程;3) 枯草芽胞杆菌是革兰氏阳性细菌,其细胞膜不均有内毒素,对人体无害,是一种公认的食品级微生物。
本文以枯草芽胞杆菌为宿主菌,对DPE基因进行了表达。由于DPE基因在组成型表达启动子HpaⅡ的调控之下,故不需要外源诱导物的加入。如图1所示,与空载体对照,有明显的目的蛋白条带 (33 kDa),且有大量的可溶性蛋白。

图1 DPE基因在B. subtilis中表达的SDS-PAGE结果Fig. 1 SDS-PAGE analysis of DPE expressed in B. subtilis. M: protein marker; 1: total cell lysate obtained from a non-expression control of B. subtilis transformed with pMA5; 2: total cell lysate obtained from B. subtilis transformed with pMA5-DPE; 3: the soluble supernatant of B. subtilis transformed with pMA5; 4: the soluble supernatant of B. subtilis transformed with pMA5-DPE.
重组DPE蛋白经过His TrapHP亲和色谱以及Source15Q阴离子交换色谱之后,纯度达到了电泳纯,如图2所示。重组DPE蛋白的理论分子量约 33 kDa,聚丙烯酰胺凝胶电泳 (12% SDS-PAGE) 的结果显示,目的蛋白条带大小符合预期。
2.2 L-RhI基因的表达与纯化
将空载体pET-21a (+) 转化到E. coli BL21中,作为对照与B. subtilis pET-21a-L-RhI同批培养并诱导表达。由图 3可知,经 IPTG诱导,pET-21a-L-RhI有明显的目的蛋白条带 (49 kDa)。将菌体破碎后,分别取上清及沉淀进行蛋白电泳后发现,表达产物大量为可溶性蛋白,包涵体较少。
重组L-RhI蛋白经过His TrapHP亲和色谱以及DEAE Sepharose Fast Flow阴离子交换色谱之后,纯度达到了电泳纯,如图4所示。重组DPE蛋白的理论分子量约49 kDa,聚丙烯酰胺凝胶电泳 (12% SDS-PAGE) 的结果显示,目的蛋白条带大小符合预期。
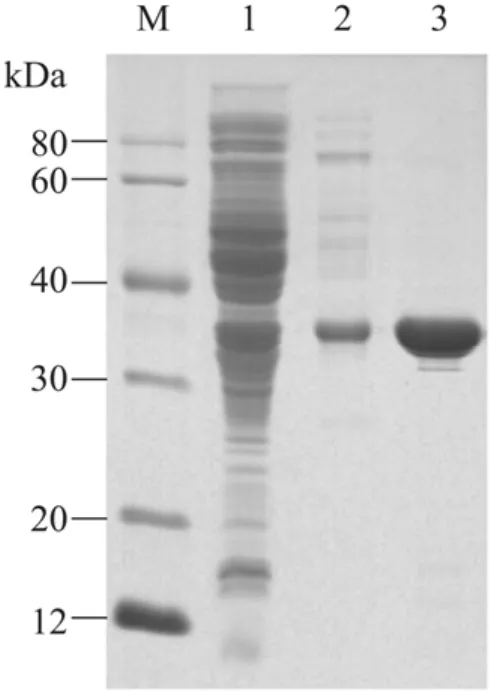
图2 重组DPE蛋白的纯化Fig. 2 Purification of the recombinant DPE expressed in B. subtilis. M: protein marker; 1: the soluble supernatant of B. subtilis transformed with pMA5-DPE; 2: recombinant DPE purified through Ni column; 3: recombinant DPE purified through Source 15Q column.

图3 L-RhI基因在E. coli中表达的SDS-PAGE结果Fig. 3 SDS-PAGE analysis of DPE expressed in E. coli. M: protein marker; 1: total cell lysate obtained from a non-expression control of E. coli BL21 transformed with pET-21a (+); 2: total cell lysate obtained from E. coli BL21 transformed with pET-21a-L-RhI; 3: the soluble supernatant of E. coli BL21 transformed with pET-21a-L-RhI; 4: the inclusion body of E. coli BL21 transformed with pET-21a-L-RhI.
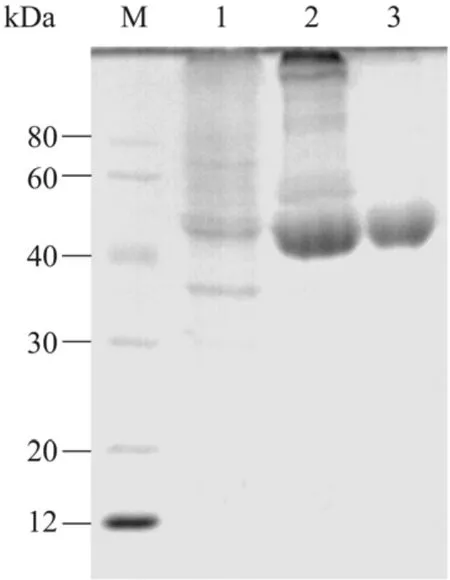
图4 重组L-RhI蛋白的纯化Fig. 4 Purification of the recombinant L-RhI expressed in E. coli. M: protein markers; 1: the soluble supernatant of E. coli transformed with pET-21a-L-RhI; 2: recombinant L-RhI purified through Ni column; 3: recombinant L-RhI purified through DEAE Sepharose Fast Flow column.
2.3 以D-果糖为底物催化生产D-阿洛糖
D-阿洛酮糖是 D-果糖的 C-3差相异构体。D-阿洛糖与D-阿洛酮糖互为醛酮异构体。从D-果糖到 D-阿洛糖的转化,需经过两步异构化反应 (图5)。自从2002年Izumoring策略揭示出了以廉价的果糖生产 D-阿洛糖的可能性,许多的研究都致力于找到合适的异构酶来完成这两步转化反应。

图5 D-果糖、D-阿洛酮糖与 D-阿洛糖间的转化方程式Fig. 5 Reaction scheme among D-fructose, D-psicose and D-allose.
2.3.1 DPE催化D-果糖生产D-阿洛酮糖
来源于C. cellulolyticum H10的DPE具备催化生产D-阿洛酮糖的催化活力 (图6)。在50 ℃反应1 h后,反应即达到了平衡。此时,D-果糖的转化率为 27.34%。本文还试验了大批量地生产D-阿洛酮糖,在底物溶液中加入了500 g/L的 D-果糖,反应的最终转化率也可以达到24.83%。
实验证明,重组C. cellulolyticum DPE适用于 D-阿洛酮糖的生产。其一,其催化反应速率高,能短时间达到反应平衡;其二,其表达宿主菌是枯草芽胞杆菌,符合食品生产的安全需求。
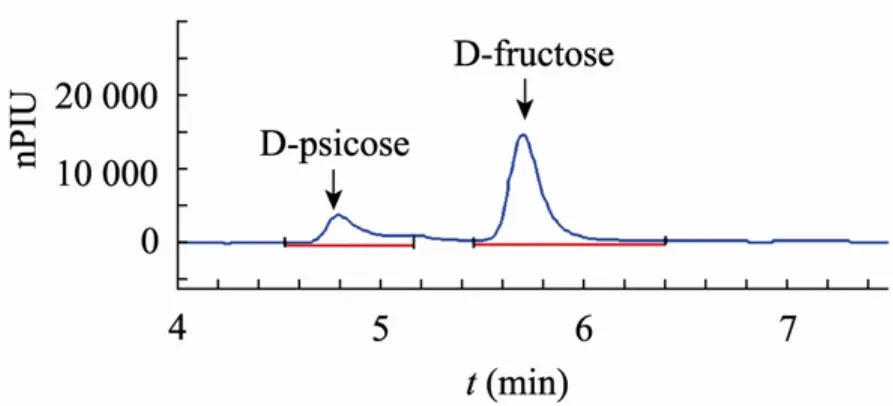
图6 以D-果糖为底物反应1 h后反应液的液相色谱图Fig. 6 HPLC of the reactants after 1 hour’s reaction using D-fructose as the substrate.
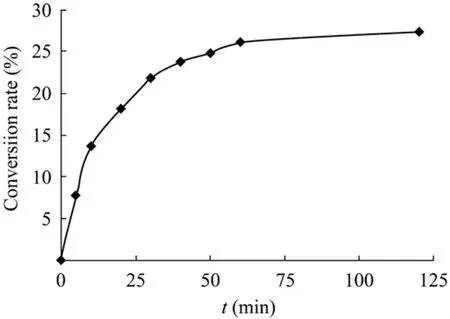
图7 催化D-果糖生成D-阿洛酮糖的反应过程曲线Fig. 7 Bioconversion of D-fructose into D-psicose using the purified DPE.
2.3.2 L-RhI催化D-阿洛酮糖生产D-阿洛糖
L-RhI是一种醛酮异构酸,它可以催化D-阿洛酮糖生产D-阿洛糖。但不是所有来源的L-RhI都具备这种催化活力,它的最适底物是 L-鼠李糖。目前为止,以D-阿洛酮糖为底物生产D-阿洛糖的L-RhI都源自假单胞菌P. stutzeri。
来源于 B. subtilis 168的 L-RhI (GenBank Accession No. AL009126.3) 与P. stutzeri L-RhI (GenBank Accession No. AB121136.1) 的氨基序列相似度不高,只有 17.23%。然而,实验结果发现,B. subtilis 168 L-RhI具有一定的催化生产D-阿洛糖的能力。如图 8所示,在 60 ℃反应24 h后,有明显的产物峰D-allose生成,此时并无其他明显的产物峰,D-psicose的转化率为34.64%。反应48 h后达到了平衡,此时底物转化率为37.51% (图9)。

图8 以D-阿洛酮糖反应24 h后反应液的液相图Fig. 8 HPLC of the reactants after 24hs’ reaction using D-psicose as the substrate.
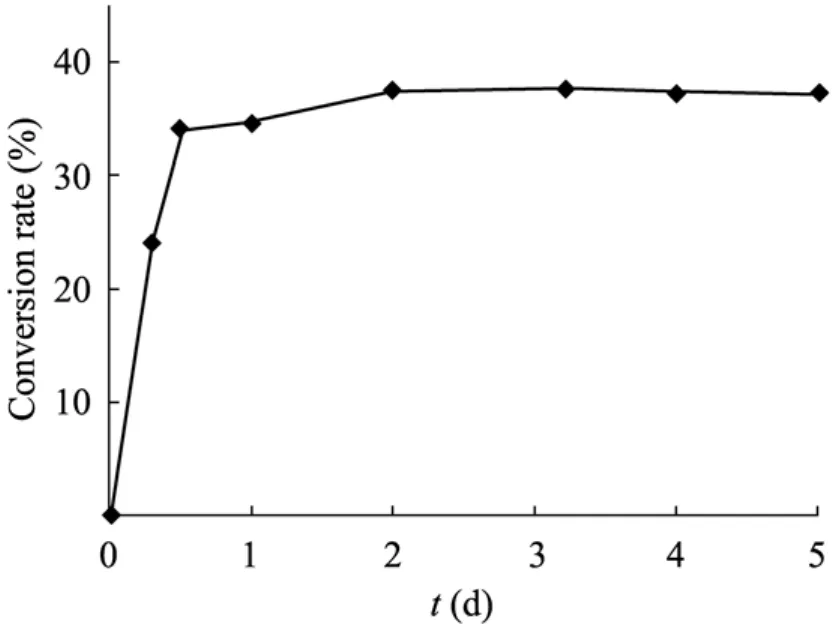
图9 催化 D-阿洛酮糖生成 D-阿洛糖的反应过程曲线Fig. 9 Bioconversion of D-psicose into D-allose using the purified L-RhI.
3 结论
本研究将来源于 C. cellulolyticum H10的DPE基因克隆到了宿主菌B. subtilis中,没有添加任何的外源诱导物,即实现了重组 DPE蛋白的高效表达。表达后的重组 DPE蛋白经过镍亲和层析和阴离子交换色谱等方法纯化,纯度达到了电泳纯。纯化后的DPE能高效地催化D-果糖转化为D-阿洛酮糖,反应1 h即可达到转化率27.34%的反应平衡。
研究还克隆了来源于Bacillus subtilis 168的L-RhI基因,并使其在宿主菌E. coli BL21 (DE3)中得到了表达。纯化后的重组L-RhI达到了电泳纯,并具有催化D-阿洛酮糖生成D-阿洛糖的催化活力。
实验证明,利用DPE和L-RhI可以将来源丰富的D-果糖经过D-阿洛酮糖转化生成D-阿洛糖,实现了稀少糖的生物转化。
[1] Izumori K. Bioproduction strategies for rare hexose sugars. Naturwissenschaften, 2002, 89(3): 120−124.
[2] Matsuo T, Suzuki H, Hashiguchi M, et al. D-psicose is a rare sugar that provides no energy to growing rats. Nutritional Vitamino, 2002, 48(1): 77. [3] Matsuo T, Baba Y,Hashiguchi M,et a1.Dietary D-psicose,a C-3 epimer of D-fructose,suppresses the activity of hepatic lipogenic enzymes in rats.Asia Pac J Clin Nutr,2001,10(3):233−237. [4] Matsuo T,Izumori K.Effects of dietary D-psicose on diurnal variation in plasma glucose and insulin concentrations of rats. Biosci Biotechnol Biochem,2006,70(9):2081−2085
[5] Murata A, Sekiya K, Watanabe Y, et al. A novel inhibitory effect of D-allose on production of reactive oxygen species from neutrophils. J Biosci Bioeng, 2003, 96(1): 89−91.
[6] Izumori K. Izumoring: a strategy for bioproduction of all hexoses. J Biotechnol, 2006, 124(4): 717−722.
[7] Ishida Y, Kamiya T, Izumori K. Production of D-tagatose 3-epimerase of Pseudomonas cichorii ST-24 using recombinant Escherichia coli. J Ferment Bioeng, 1997, 84(4): 348−350.
[8] Kim NH, Kim HJ, Kang DI, et al. Conversion shift of D-fructose to D-psicose for enzyme-catalyzed epimerization by addition of borate. Appl Environ Microb, 2008, 74(10): 3008−3013.
[9] Takeshita K, Suga A, Takata G, et al. Mass production of d-psicose from d-fructose by a continuous bioreactor system using immobilized d-tagatose 3-epimerase. J Biosci Bioeng, 2000, 90(4): 453−455.
[10] Bhuiyan SH, Itami Y, Rokui Y, et al. D-allose production from d-psicose using immobilized l-rhamnose isomerase. J Ferment Bioeng, 1998, 85(5): 539−541.
[11] Korndörfer IP, Fessner WD, Matthews BW. The structure of rhamnose isomerase from Escherichia coli and its relation with xylose isomerase illustrates a change between inter and intra-subunit complementation during evolution. J Mol Biol, 2000, 300(4): 917−933.
[12] Doan TNT, Prabhu P, Kim JK, et al. Crystallization and preliminary X-ray crystallographic analysis of l-rhamnose isomerase with a novel high thermostability from Bacillus halodurans. Acta Cryst, 2010, F66(6): 677−680.
[13] Park CS, Yeom SJ, Lim YR, et al. Characterization of a recombinant thermostable l-rhamnose isomerase from Thermotoga maritima ATCC 43589 and its application in the production of l-lyxose and l-mannose. Biotechnol Lett, 2010, 32(12): 1947−1953.
[14] Mu WM, Chu FF, Xing QC, et al. Cloning, expression, and characterization of a D-psicose 3-epimerase from Clostridium cellulolyticum H10. J Agric Food Chem, 59(14): 7785−7792.
[15] Zhang LT, Mu WM, Jiang B, et al. Characterization of d-tagatose-3-epimerase from Rhodobacter sphaeroides that converts d-fructose into d-psicose. Biotechnol Lett, 2009, 31(6): 857−862.
猜你喜欢
云南化工(2021年6期)2021-12-21 07:30:56
中成药(2021年5期)2021-07-21 08:39:12
中国科技纵横(2021年24期)2021-03-02 06:42:52
科学(2020年2期)2020-08-24 07:57:00
食品界(2019年8期)2019-10-07 12:27:36
中成药(2018年6期)2018-07-11 03:01:12
中成药(2016年4期)2016-05-17 06:07:50
生物技术通报(2015年1期)2015-04-10 16:15:19
山西大同大学学报(自然科学版)(2013年5期)2013-09-13 10:44:14
化学分析计量(2013年3期)2013-03-11 16:37:32
