
斜卧青霉去泛素化蛋白酶CREB的缺失提高纤维素酶的生产
2012-02-10 01:20周广麒吕晶李忠海李晶晶王明钰曲音波肖林覃树林赵海涛夏蕊蕊方诩
生物工程学报 2012年8期
周广麒,吕晶,李忠海,李晶晶,王明钰,曲音波,肖林,覃树林,赵海涛,夏蕊蕊,方诩
1 大连工业大学生物工程学院,辽宁 大连 116034
2 山东大学微生物技术国家重点实验室,山东 济南 250100
3 山东大学药学院,山东 济南 250012
4 山东龙力生物科技股份有限公司,山东 德州 251200
5 山东省秸秆生物炼制技术企业重点实验室,山东 德州 251200
木质纤维素是自然界中分布最为广泛的可再生资源。利用木质纤维素生产洁净的生物能源是将来解决石化产品污染的重要举措。丝状真菌能产生多种纤维素酶和半纤维素酶并有效降解木质纤维素生成多用途糖类。斜卧青霉Penicillium decumbens T.[1-3]作为重要的高产纤维素酶丝状真菌之一,已应用于纤维素酶及半纤维素酶的工业生产,但对于高效低成本地降解利用木质纤维素,还需要对该菌株的多种代谢途径进行系统的菌株改造。
微生物细胞中,碳源代谢阻遏作为重要的生理代谢控制机制使细胞在较容易利用的碳源存在时,相比其他较难代谢的碳源具有优先利用的特点。丝状真菌中参与碳源代谢阻遏的蛋白主要有CREA (CRE1)[4]、CREB[5]、CREC[6]及CRED[7]。CREA 与酿酒酵母 Saccharomyces cerevisiae H.中的 Mig1/Mig2/Mig3蛋白同源[8],为重要的转录调控因子,介导葡萄糖在碳源代谢阻遏中的作用[9],同时,CREA蛋白可与泛素结合,形成泛素化蛋白[7]。泛素化的CREA相对不稳定,容易受到蛋白酶的降解。CREB和CREC分别编码1个去泛素化蛋白酶[5],在多核真核细胞中较为普遍,但在单核细胞酿酒酵母中未发现同源蛋白的存在。在构巢曲霉Aspergillus nidulans W.中,creB基因编码1个由767个氨基酸,包括6个去泛素化结构域组成的去泛素化酶,属于泛素特异性加工酶 (Ubiquitin-specific processing enzymes,UBP) 家族中的一员,其中的卷曲螺旋结构域起到底物识别的作用[9]。CreB是第1个被发现参与碳代谢阻遏作用的去泛素化酶,CreB在第 240位、385位、473位和538位的氨基酸有4个重要的PEST序列,PEST序列是一段富含脯氨酸(P)、谷氨酰胺 (E)、丝氨酸 (S)、苏氨酸 (T),约由 10个氨基酸组成的结构序列,被认为是泛素化识别序列之一[10]。CreB的同源序列大部分存在比较高等的生物中,比如人类的UBH1,拟南芥的UBP3,毕赤酵母Pichia pastoris Yeast C.中的UBP1以及黑腹果蝇的AAF56066。在构巢曲霉中 CREB的缺失可以引起多种纤维素酶的产量提高[5],Denton等在里氏木霉 Trichoderma reesei中敲除creB的同源序列cre2,同样可以达到提高纤维素酶产量的作用[11]。CREB和CREC可形成去泛素化复合体,共同作用于泛素化的蛋白质并使其去泛素化,从而使去泛素化的蛋白质更为稳定[6]。CRED包含1个抑制蛋白结构域和1个PY模块与酿酒酵母中的Rod1p和Rog3p蛋白具有高度的相似性,creD参与 1个与CREB-CREC复合物的去泛素化作用完全相反的过程,可以对靶蛋白进行泛素化标记[7]。由于蛋白质 (如 CREA) 泛素化水平的增高会造成该蛋白在细胞中受降解程度的变化,进而影响到细胞利用碳源的代谢途径,因此,CREB的缺失对真菌利用纤维素作为碳源分泌纤维素酶的影响值得深入研究。
通过敲除与构巢曲霉creB基因同源的基因,获得了该基因的突变株,研究CREB在斜卧青霉利用纤维素时对产纤维素酶的影响,对该突变株的表型,产纤维素酶及产胞外蛋白的能力进行了测定和分析。
1 材料与方法
1.1 材料
1.1.1 菌株和质粒
斜卧青霉Ku-39 (Δpku70::hph)、菌株Kup-1 (Δpku70::hph;ΔpyrG::ptrA) 以及质粒 pEKU由山东大学微生物技术国家重点实验室保存,pMD18-T载体购自 TaKaRa公司,大肠杆菌DH5α感受态细胞购自TransGen Biotech公司。
1.1.2 培养基
麸皮培养基:100 g麸皮加 1 L水,煮沸30 min,过滤后定容至1 L。固体麸皮培养基加2%琼脂粉。LB培养基和测生物量液体培养基分别参照文献[12]和[13]。表型分析培养基:Mandel′s营养盐液 (1倍),1‰ Triton-100,2%琼脂粉,碳源分别为2%葡萄糖,1%微晶纤维素,1%葡萄糖+1%微晶纤维素,2%可溶性淀粉,pH 5.5。测酶活培养基:Mandel′s 营养盐液(1倍),1%微晶纤维素,pH 5.5。
1.1.3 主要试剂
Taq、FastPfu DNA 聚合酶购自 TransGen Biotech公司、限制性内切酶购自Fermentas公司,琼脂糖凝胶/PCR产物纯化试剂盒购自 Biomiga公司,细胞壁裂解酶 (L1412-25G) 购自 Sigma公司,改良型Bradford法蛋白质浓度测定试剂盒购自上海生工生物工程技术服务有限公司,Southern杂交试剂盒购自罗氏诊断产品 (上海)有限公司,pMD18-T载体购自TaKaRa公司。
1.2 方法
1.2.1 斜卧青霉分子生物学操作方法
质粒提取:碱裂解法[12],将带有质粒的大肠杆菌培养14 h,离心,倒掉上清液,加入溶液Ⅰ(GET) 充分悬浮菌体,再加入溶液Ⅱ (变性液),加盖颠倒6~7次充分混匀,加入溶液Ⅲ颠倒混合均匀后冰上放置 5 min,离心将上清液转移到1.5 mL离心管中,利用醇沉的方法得到沉淀,用适量双蒸水溶解。
真菌转化:斜卧青霉Kup-1的转化通过细胞壁裂解酶消化细胞壁得到原生质体,再利用PEG-CaCl2法转化目的DNA片段[13]。
染色体DNA提取:真菌染色体DNA的提取采用液氮研磨法[13]。接种浓度为1×107个/mL新鲜的分生孢子于液体培养基中,30 ℃、200 r/min培养48 h,将菌体过滤收集,放入研钵中加入液氮进行研磨破碎,收集破碎后的菌体于15 mL离心管中加入适量抽提缓冲液,65 ℃水浴,然后加入苯酚/氯仿振荡充分混合均匀后,离心收集上清液,加入NaAc和异丙醇,−20 ℃放置20 min后离心收集沉淀物,用70%乙醇,洗涤1次,待乙醇完全挥发后加入双蒸水溶解。
1.2.2 转化子表型分析
分别吸取1 μL浓度为1×105个/mL的孢子悬液,点接于表型分析培养基平板,30 ℃ 静置培养9 d。
1.2.3 蛋白质含量测定
参考改良型 Bradford法蛋白质浓度测定试剂盒说明。
1.2.4 生物量测定
接 1×107个/mL分生孢子悬液 100 μL至50 mL液体基本培养基中,30 ℃、200 r/min 培养,分别在24、33、42、51、60、69 h时取样,真空过滤抽干,80 ℃烘干24 h,称重。
1.2.5 滤纸酶活、木聚糖酶活、内切葡聚糖酶活、外切葡聚糖酶活力的测定
滤纸酶活:1 cm×6 cm定量滤纸条,1 mL醋酸缓冲液 (pH 4.8) 和0.5 mL稀释后的粗酶液,50 ℃酶解1 h。
木聚糖酶活:1 mL 1%的燕麦木聚糖悬浮液,0.5 mL稀释后的酶液,50 ℃酶解30 min。
内切葡聚糖酶活:1 mL 1%的CMC-Na溶液,0.5 mL稀释后的酶液,50 ℃酶解30 min。
以上3种酶活测定中,均以DNS法测定酶解液中的还原糖量。
外切葡聚糖酶活:0.5 mL稀释后的粗酶液,加入50 µL pNPC (g/L),50 ℃保温30 min;加入150 µL 10% NaCO3终止反应[14]。
酶活力单位:1分钟内水解底物产生1 μmol还原糖或对硝基苯酚所需的酶量定义为1个酶活力单位 (IU)。
1.2.6 pH测定
取 1 mL产酶发酵液,12 000 r/min离心3 min,吸取上清液,测pH值。
1.2.7 ΔcreB::pyrG敲除盒的构建
ΔcreB::pyrG 敲除盒的构建 (图 1)。ΔcreB::pyrG敲除盒上、下游同源臂 (5′-flanking,3′-flanking) 以及上游同源臂部分重复序列(5′-flanking RE) 的扩增均以Ku-39染色体DNA为模板,分别以引物对 CreB-uF+CreB-uR;CreB-dF+CreB-dR1和CreB-reF+CreB-reR为引物(表1)。pyrG筛选标记表达盒的扩增以质粒pEKU (山东大学微生物技术国家重点实验室保存) 为模板,PyrG-F1和PyrG-F2为引物。ΔcreB::pyrG敲除盒下游 3个片段的连接采用 Double-joint PCR[15]方法,融合 pyrG 筛选标记表达盒、ΔcreB::pyrG敲除盒上游同源臂部分重复序列以及下游同源臂 3个片段,再以 PyrG-F2和CreB-dR2为嵌套引物,PCR扩增获得融合产物,并将该融合产物经琼脂糖凝胶纯化回收后经T/A连接插入到克隆载体pMD18-T上,获得重组质粒pCreB-DT,转化DH5α感受态细胞。将质粒pCreB-DT和ΔcreB::pyrG敲除盒上游同源臂分别进行XbaⅠ和SmaⅠ双酶切,然后通过T4 DNA连接酶进行连接,获得重组质粒 pCreBqch,带有上游部分同源臂序列的ΔcreB::pyrG 敲除盒构建成功。将重组质粒pCreBqch转化DH5α感受态细胞。以质粒 pCreBqch为模板,CreB-uF和CreB-dR2为引物PCR大量扩增ΔcreB::pyrG敲除盒,转化斜卧青霉Kup-1原生质体。
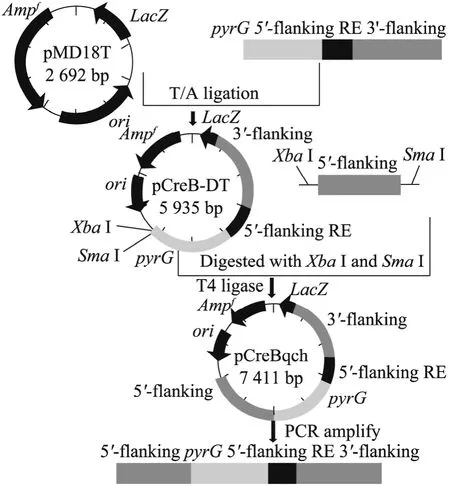
图1 ΔcreB::pyrG敲除盒构建流程图Fig. 1 Flow diagram of ΔcreB::pyrG.

表1 本研究所用引物序列Table 1 Primers used in this study
Double-joint PCR反应体系:第1轮PCR, 50 μL反应体系为:40 μL双蒸水,上下游引物各1 μL,dNTPs (10 mmol) 1 μL,模板1 μL,10×缓冲液5 μL,Taq DNA聚合酶1 μL。PCR反应条件为:94 ℃预变性2 min;94 ℃, 20 s;58 ℃, 30 s;72 ℃, 1 min 30 s,32个循环;72 ℃延伸10 min。分别切胶回收3个第1轮PCR产物:上游同源臂、下游同源臂以及上游同源臂部分重复序列,来进行第2轮PCR,本轮PCR不需要添加引物,反应体系为:40 μL双蒸水,dNTPs (10 mmol) 1 μL,3个DNA片段各1 μL,10×缓冲液5 μL,HIFI酶1 μL。反应条件为:94 ℃预变性2 min;94 ℃, 20 s; 55 ℃, 10 min;72 ℃, 4 min,12个循环;72 ℃延伸10 min。将第2轮PCR产物适当稀释作为第3轮PCR的模板;40 μL双蒸水,dNTPs (10 mmol) 1 μL,10×缓冲液5 μL,HIFI酶1 μL。反应条件为:94 ℃预变性2 min;94 ℃, 20 s;55 ℃, 30 s;72 ℃, 5 min,32个循环;72 ℃延伸10 min。
斜卧青霉creB基因序列已提交 GenBank,登录号为JN977580。
1.2.8 系统进化树分析
根据斜卧青霉中 CreB氨基酸序列,自NCBI (National Center for Biotechnology Information)数据库比对筛选出该蛋白在丝状真菌中的同源序列。CreB同源序列分别来自产黄青霉Penicillium chrysogenum T.、构巢曲霉A. nidulan、烟曲霉Aspergillus fumigates、黑曲霉Aspergillus niger、米曲霉 Aspergillus oryzae、Talaromyces stipitatus、马尔尼菲青霉菌Penicillium marneffei、里氏木霉 T. reesei、粗糙脉孢菌 Neurospora crassa。邻接系统发育树 (Neighbor Joining tree)采用软件包ClustalX 2.0和MEGA4.1分析构建,标尺为每位点氨基酸替代值。
2 结果与分析
2.1 ΔcreB::pyrG敲除盒的构建
为得到 creB基因缺失突变株,首先构建了ΔcreB::pyrG 敲除盒。首先,分别扩增得到了ΔcreB::pyrG敲除盒上游同源臂序列1 476 bp、pyrG筛选标记表达盒1 538 bp、ΔcreB::pyrG敲除盒上游同源臂部分重复序列 531 bp以及ΔcreB::pyrG敲除盒下游同源臂序列 1 733 bp (图2泳道1–4)。采用Double-joint PCR[15]的方法将pyrG筛选标记表达盒、ΔcreB::pyrG敲除盒上游同源臂部分重复序列以及ΔcreB::pyrG敲除盒下游同源臂,3个片段融合,嵌套引物PyrG-F2+ CreB-dR2扩增得到3片段融合产物 (图2泳道5),然后将该融合片段连接到pMD18-T载体上,得到重组质粒 pCreB-DT (图 2泳道 6)。将质粒pCreB-DT和 ΔcreB::pyrG敲除盒上游同源臂(图2泳道6, 1) 分别进行XbaⅠ和SmaⅠ双酶切,酶切产物经切胶回收后通过 T4 DNA连接酶连接,得到重组质粒 pCreBqch (图 2泳道9)。以pCreBqch为模板,CreB-uF和CreB-dR2为引物扩增ΔcreB::pyrG敲除盒 (图2泳道13)。Hind Ⅲ和KpnⅠ分别单酶切验证ΔcreB::pyrG敲除盒,经Hind Ⅲ酶切后电泳结果显示得到3条带大小分别为725 bp、1 185 bp以及2 819 bp (图2泳道14),KpnⅠ酶切后电泳结果显示得到3条带大小分别为482 bp、1 927 bp以及2 320 bp (图2泳道15),2个酶切结果均与预期目的条带大小一致,表明ΔcreB::pyrG敲除盒构建成功。
重组质粒 pCreB-DT由华大基因进行测序验证。
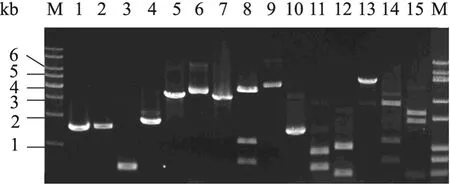
图2 ΔcreB::pyrG敲除盒的构建以及酶切验证Fig. 2 Construction of creB gene deletion cassette and the confirmation of the cassette structure by restriction enzyme digestion. 1: 5'-flanking region of creB gene in the deletion cassette; 2: pyrG; 3: Partial repeating sequence of the 5'-flanking region of creB gene in the deletion cassette; 4: 3'-flanking region of creB gene in the deletion cassette; 5: the fused cassette of 2,3,4 using nest PCR; 6: pCreB-DT vector; 7: amplification of the fused cassette of 2,3,4 using nest PCR from 6; 8: pCreB-DT vector digested with Hind Ⅲ; 9: pCreBqch; 10: colony PCR confirmation of pCreBqch; 11: 10 digested by EcoR I; 12: 10 digested by Kpn I; 13: the creB gene deletion cassette; 14: 13 digested by Hind III; 15: 13 digested by Kpn I.
2.2 ΔcreB突变株的构建
将构建成功的 ΔcreB::pyrG 敲除盒转入Kup-1菌株中。在转化平板上获取转化子10~15个/μg DNA。为排除转化子中的异核体,挑取转化子在平板上经过2轮划线分单孢复筛培养。将纯化后的转化子接入到基本培养基中,30 ℃、200 r/min摇床培养 48 h。提取转化子基因组DNA。以提取的基因组 DNA为模板,PyrG-F2和PyrG-R为引物扩增pyrG表达盒部分序列,(图3,泳道1–2),扩增出大小为1 345 bp的目的条带,表明 ΔcreB::pyrG敲除盒已成功转入到出发菌株中。CreB-YZF1和CreB-YZR1 (序列见表 1) 为引物,其中引物 CreB-YZF1和CreB-YZR1结合位点分别位于ΔcreB::pyrG敲除盒上游同源臂和下游同源臂,分别以出发菌株及转化子基因组DNA为模板,出发菌株扩增获得3 573 bp大小目的条带 (图3,泳道3),而转化子扩增出片段大小为 2 652 bp大小目的条带(图 3,泳道 4–5),表明 ΔcreB::pyrG敲除盒在creB基因位点发生同源双交换,creB基因编码区已被置换;为进一步验证creB基因已被同源敲除,以PyrG-YF和CreB-dR1为引物,分别以出发菌株及2株不同转化子基因组DNA为模板,进行PCR扩增。其中引物PyrG-YF和CreB-dR1的结合位点分别位于 pyrG基因编码区和ΔcreB::pyrG 敲除盒下游同源臂外侧的染色体上。由于出发菌株不能扩增出相应大小的目的条带 (图3,泳道6),而以2株转化子DNA为模板,上下游引物可配对扩增出大小为2 515 bp的目的条带 (图3,泳道7–8) 说明2株转化子发生同源双交换。以上结果表明,ΔcreB突变株已构建成功。
2.3 ΔcreB突变株的Southern blotting分析
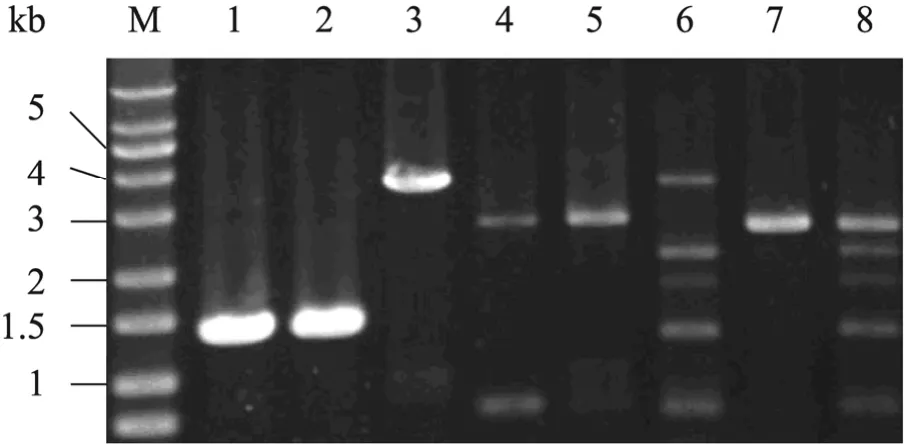
图3 ΔcreB转化子的PCR验证Fig. 3 Verification of transformants by PCR analysis. 1–2: amplification of pyrG gene with genomic DNA of the transformants; 4–5, 7–8: amplification was performed with genomic DNA of the primary transormants; 3, 6: amplification was performed with genomic DNA of ΔpyrG::ptrA.
为分析ΔcreB::pyrG敲除盒在转化子染色体中的整合类型,对得到的转化子进行 Southern blotting (图 4A) 检测,首先分别提取出发菌株Kup-1及 ΔcreB::pyrG 敲除盒转化子基因组DNA,然后经限制性内切酶 BamHⅠ酶切,ΔcreB::pyrG敲除盒下游同源臂 DNA片段为探针进行 Southern杂交 (图 4B)。杂交结果显示ΔcreB突变株出现单1条带 (图4A,泳道1),该条带大小符合 ΔcreB::pyrG敲除盒在 creB位点以同源双交换整合方式所产生杂交片段的大小,并未出现出发菌株杂交所示条带 (图 4A WT)。由于ΔcreB突变株未出现因敲除盒的随机插入而产生的条带,因此,杂交结果表明ΔcreB::pyrG敲除盒是以单拷贝的形式整合到转化子染色体上。
2.4 ΔcreB突变株的表型分析
为分析creB基因缺失对该菌株表型的影响,吸取ΔcreB突变株及菌株Ku-39分生孢子悬液,分别点接于葡萄糖、微晶纤维素、葡萄糖+微晶纤维素以及可溶性淀粉为碳源的平板。在以葡萄糖为唯一碳源的基本培养基上,ΔcreB突变株的菌落形态以及生长速率相比菌株 Ku-39均未发生明显变化 (图5A),表明creB基因的缺失对菌株的营养生长未产生明显的影响。在以可溶性淀粉为唯一碳源的平板上,ΔcreB突变株的菌落生长表型相比Ku-39菌株也无明显变化,但平板经碘化钾染色后 (图 5D),突变株所形成的淀粉水解圈变大,表明ΔcreB突变株分泌淀粉酶的活性增强。在以微晶纤维素为唯一碳源的培养基上(图5B),ΔcreB突变株生长正常,与对照菌株相比无明显差异,但在菌株ΔcreB菌落周围出现明显的因纤维素降解所形成的透明圈,而菌株Ku-39菌落周围未出现纤维素水解透明圈,表明creB基因的缺失可提高突变株产纤维素酶的活性,特别在葡萄糖存在的培养条件下,以微晶纤维素+葡萄糖为复合碳源的培养基上 (图 5C),ΔcreB突变株仍能形成较为明显的纤维素水解透明圈,表明 creB基因的缺失不仅能提高产纤维素酶的能力,而且还呈现出一定的抗葡萄糖代谢阻遏效应。
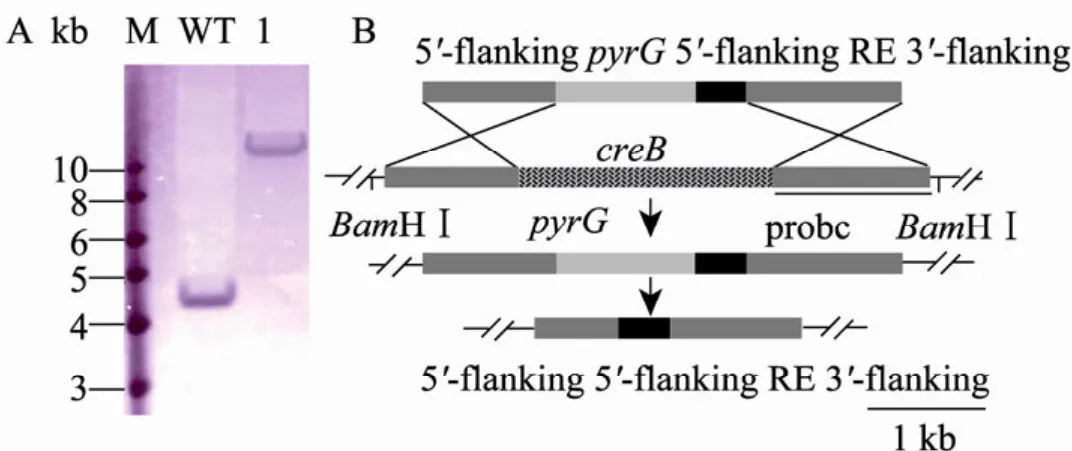
图4 斜卧青霉Kup-1 creB敲除Southern blotting分析Fig. 4 Southern blotting analysis of the P. decumbens Kup-1 ΔcreB knockout strain. (A) Southern blotting analysis of the knockout strain. M: 1 kb ladder. WT: Southern blotting of genomic DNA extracted from wild-type. 1: Southern blotting of genomic DNA extracted from the knockout strain. (B) Diagram of creB deletion by homologous recombination.
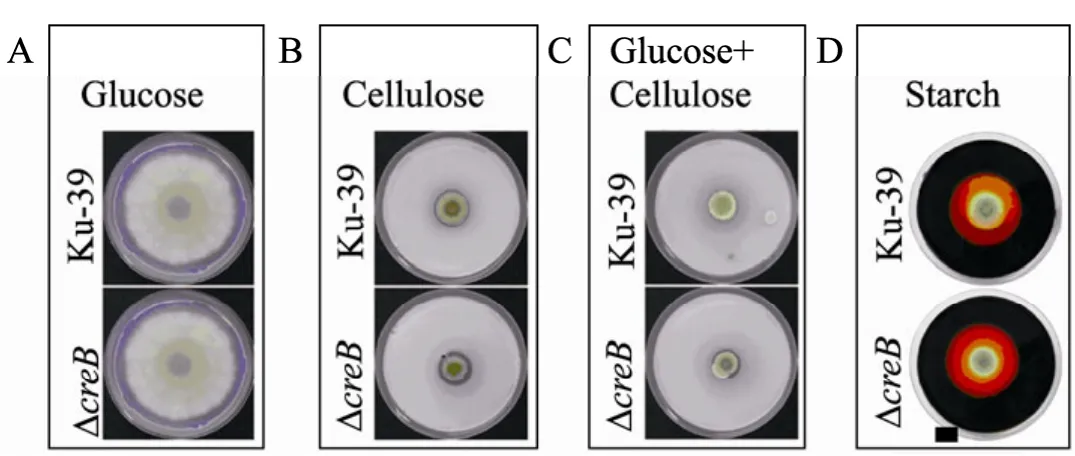
图5 ΔcreB突变株的平板检测分析Fig. 5 Comparing the morphologies of ΔcreB and the Ku-39 strain cultivated for 9 days. (A) Glucose. (B) Cellulose. (C) Glucose +Cellulose. (D) Starch.
2.5 pH值的变化
为测定 CREB的缺失对菌株在液体培养条件下发酵的影响,对菌株Ku-39和ΔcreB突变株不同培养时间段发酵液的 pH值进行了测定(图6)。ΔcreB突变株和Ku-39菌株发酵液的pH值在48 h内均明显处于下降的状态,在24 h处,ΔcreB突变株发酵液的pH值下降速度明显慢于出发菌株。Ku-39菌株和ΔcreB突变株发酵液的pH值48 h前后分别达到最低点3.6和3.5,并在48 h与120 h之间发酵液pH值稳步上升达到5.2左右,随后 pH值趋于稳定,结果表明,CREB的缺失对突变株在以微晶纤维素为唯一碳源的发酵液pH值没有明显变化。
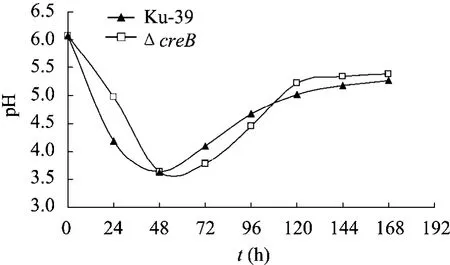
图6 菌株Ku-39和ΔcreB突变株发酵过程的pH值Fig. 6 pH value of P. decumbens Ku-39 and ΔcreB.
2.6 酶活力的变化
在液体培养产酶条件下,对菌株 Ku-39和ΔcreB突变株不同培养时间段发酵液的滤纸酶活、内切葡聚糖酶活、木聚糖酶活、外切葡聚糖酶活进行了测定 (图7)。ΔcreB突变株发酵液的滤纸酶活、内切葡聚糖酶活、木聚糖酶活、外切葡聚糖酶活均比出发菌株的相应酶活显著提高,其中,ΔcreB突变株发酵液的滤纸酶活在第144 h时,比Ku-39菌株发酵液提高了1.8倍 (图7A),内切葡聚糖酶活在96 h比出发菌株提高1.71倍(图 7B),木聚糖酶活在 120 h时提高 2.06倍(图7C),外切葡聚糖酶活在96 h时提高2.04倍(图7D)。ΔcreB突变株纤维素酶活的提高与在固体纤维素平板上该突变株的表型一致,均表现出纤维素酶分泌活性的显著增加,由此表明,CREB的缺失可显著增强突变株产纤维素酶的能力,进而提高菌株降解利用纤维素的效能。
2.7 分泌蛋白质总量变化
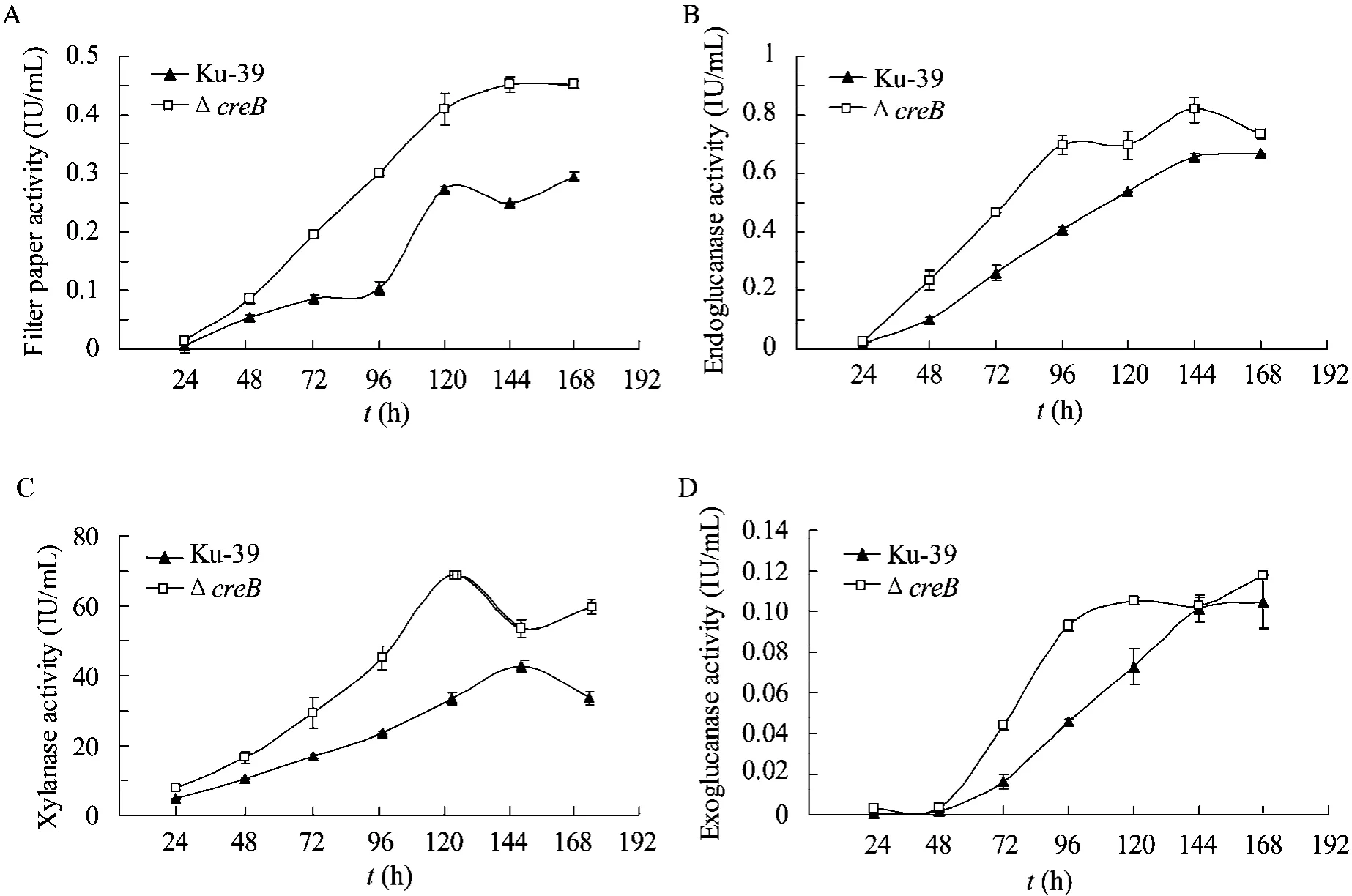
图7 菌株Ku-39和ΔcreB突变株的滤纸酶活、内切葡聚糖酶活、木聚糖酶活、外切葡聚糖酶活Fig. 7 Filter paper activity, endoglucanase activity, xylanase activity and exoglucanase activity by P. decumbens Ku-39 and ΔcreB. (A) Production of filter paper activity by P. decumbens Ku-39 and ΔcreB. (B) Production of endoglucanase activity by P. decumbens Ku-39 and ΔcreB. (C) Production of xylanase activity by P. decumbens Ku-39 and ΔcreB. (D) Production of exoglucanase activity by P. decumbens Ku-39 and ΔcreB.
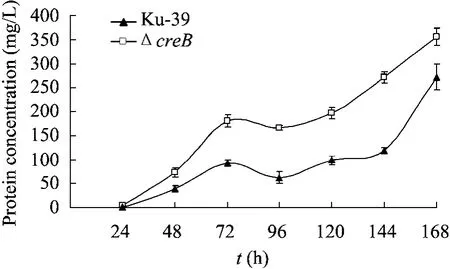
图8 菌株Ku-39和ΔcreB突变株的分泌蛋白质含量Fig. 8 Concentration of secreted protein of P. decumbens Ku-39 and ΔcreB during the fermentation.
在液体培养产酶条件下,对菌株 Ku-39和ΔcreB突变株不同培养时间段发酵上清液蛋白质含量进行了测定 (图8)。在培养至24 h时,ΔcreB突变株与菌株 Ku-39发酵液的蛋白质含量均比较低,自培养24 h开始,ΔcreB突变株发酵上清液中的蛋白质含量相比对照菌株明显提高,其中在72 h、96 h、120 h、144 h以及168 h时,其蛋白质含量分别达到菌株 Ku-39的 1.93倍、2.68倍、2.00倍、2.30倍以及1.31倍,胞外蛋白质浓度总体呈明显的上升趋势。因此,creB基因的缺失可导致细胞外蛋白质含量的显著增加。
2.8 生物量变化
为测定 creB基因的缺失是否影响菌株的营养生长,接浓度为1×107个/mL的分生孢子到液体基本培养基中。ΔcreB突变株和菌株Ku-39在培养24 h后,进入了对数生长期,并且在48 h左右达到菌体的最大量。在培养48 h后,ΔcreB突变株生物量进入生长稳定的平台期,生物量相比菌株Ku-39略低。在液体培养条件下,creB 基因的缺失可轻度影响突变株的营养生长 (图9)。
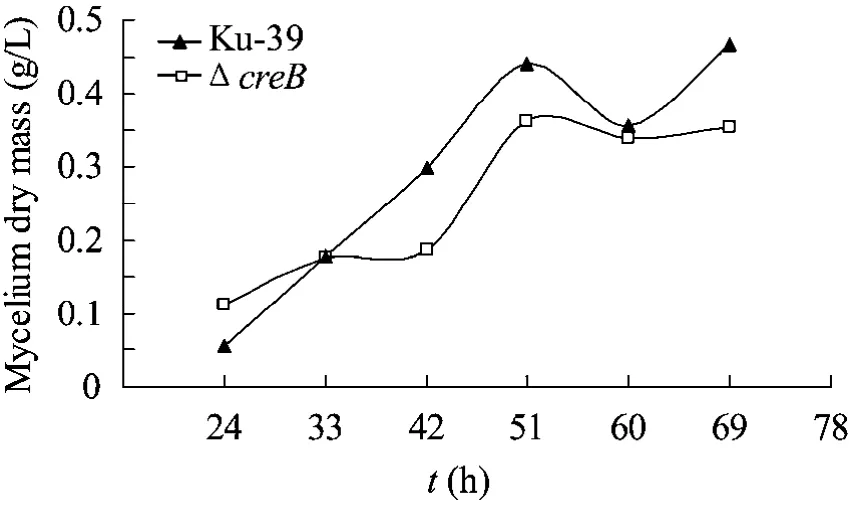
图9 菌株Ku-39和ΔcreB突变株的生长量Fig. 9 Growth of P. decumbens Ku-39 and ΔcreB in glucose-based medium.
2.9 CREB在丝状真菌中的系统进化
从CreB系统进化树 (图10) 可以发现,CreB存在于许多产纤维素酶丝状真菌中,并且其氨基酸序列与产黄青霉以及曲霉属来源的CreB同源序列有较高的相似度。因此可以推测CreB在丝状真菌纤维素酶合成过程中具有重要的作用。
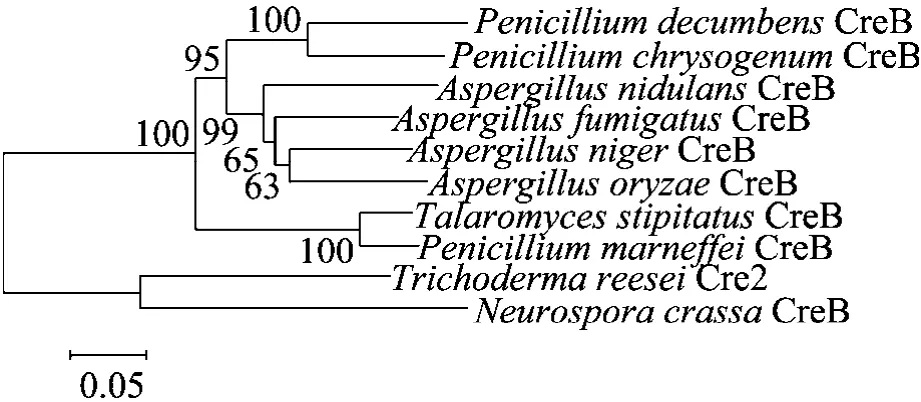
图10 CreB系统进化树Fig. 10 The neighbor joining tree of CreB. Deubiquitinating enzyme CreB are highly conserved across most sequenced filamentous fungi. The neighbor joining tree above shows the phylogenetic relationship of some of the fungal CreB in the NCBI protein database and their relationship to P. decumbens CreB. The bootstrap values are shown and the The bar=0.05 represents genetic distance in substitutions per amino acid.
3 讨论
通过同源双交换整合的方式在斜卧青霉中敲除 creB基因,获得了产纤维素酶和胞外蛋白质含量均提高的突变株ΔcreB。creB基因编码1种去蛋白质泛素化酶[5],因此,开展微生物蛋白质的泛素化与去泛素化的研究对于菌株的改造具有重要的意义。
真菌遗传转化系统中可利用的筛选标记比较有限。现在较为常用的筛选标记主要有ptra[16]、hph[17]、Hyg[18]、pyrG[19]和amdS[20]等,但工业菌株的系统改造需要在同一株菌中进行多次的遗传操作,因此,需要在斜卧青霉中通过构建可重复利用的筛选标记操作系统,用于该菌累积式的系统改造。在斜卧青霉中已经构建了高效的遗传基因打靶系统菌株Ku-39[21],并在此基础上获取了 pyrG缺失突变株 Kup-1 (数据未发表)。在构建creB敲除盒的过程中,在该敲除盒的pyrG表达盒两侧加入同源臂部分重复序列。由于该菌株具有高效的同源重组效率,因此,在含有 5′-氟乳清酸 (5′-FOA) 的平板上可高效筛选pyrG筛选标记自我剪切的菌株。筛选标记重复利用系统的建立为该菌株代谢途径的优化重组,进而为构建高产纤维素酶菌株提供了良好的基因操作基础。
蛋白质的泛素化可导致蛋白质的降解和影响蛋白质的活性。ΔcreB突变株也可在含有葡萄糖的纤维素平板上形成透明圈,表明该突变株在葡萄糖存在的情况下仍有较强的纤维素酶活性。在丝状真菌降解利用纤维素等过程中,如存在较容易利用的碳源,则可抑制纤维素酶的产生,从而产生碳源代谢阻遏效应。负责碳源代谢阻遏的蛋白主要有 CREA[4]、CREB[5]、CREC[6]及CRED[7]。CREA作为1种重要的转录调控因子,可通过影响其他调控因子如 XlnR[22-23],间接调控木聚糖的利用,并且,CREA的缺失可造成明显的抗代谢物阻遏作用。CREB和CREC分别编码1种去蛋白质泛素化的蛋白,这2种蛋白的结合可形成具有去蛋白质泛素化功能的复合物[6],该复合物可与泛素化后的 CREA相结合并使其去泛素化,进而增强CREA的稳定性,而CREB的缺失则造成CREC和CREB不能形成有效的去泛素化复合物,从而导致泛素化后的CREA蛋白不稳定,容易受到蛋白酶的降解,进而减少葡萄糖代谢的阻遏效应,同样,在缺失CREC蛋白的情况下,也可产生与 CREB缺失相类似的现象(数据未发表)。因此,开展对蛋白质的降解机理的研究对于提高菌株蛋白质的产量,特别是针对菌株高产工业用酶的研究具有较强的指导作用。
碳源代谢阻遏调控机制的存在,使细胞能优先利用营养价值较高的碳源作为细胞的能源,特别是在葡萄糖等较易利用碳源存在条件下,可显著降低纤维素酶的产量。丝状真菌中纤维素酶的表达转录明显受转录因子 CREA/CRE1[24-26]、XLNR/XYR1[27-29]、ACE1、ACE2[24]等的调控,其中CREA/CRE1是细胞产生碳源代谢阻遏过程中特别重要的调控中间体。在里氏木霉[25]、斜卧青霉[13]等丝状真菌中,CREA/CRE1的缺失造成纤维素酶表达过程中碳源代谢阻遏现象的降低或消除,并使相应突变株中纤维素酶表达量明显提高。里氏木霉中,XYR1和ACE1分别作为纤维素酶表达重要的正向和负向转录调控因子,CRE1的完全缺失明显降低XYR1的转录活性,并且导致ACE1的表达显著提高,表明ACE1也受到了明显的碳源代谢阻遏效应[25]。里氏木霉中ACE2可正向调控纤维素酶的表达。CRE1蛋白的完全缺失可导致ACE2表达量显著降低[25],然而在构巢曲霉、斜卧青霉和黑曲霉中,未发现与ACE2同源的调控蛋白,表明丝状真菌纤维素酶系的表达调控存在较大区别。由于CREA/CRE1的稳定性受细胞蛋白质泛素化/去泛素化机制的明显影响,因此,深入研究丝状真菌重要蛋白质泛素化的机制对于消除或降低纤维素酶表达的碳源代谢阻遏效应有重要的作用。
提高丝状真菌产纤维素酶的活性一直是重要的研究领域。通过缺失去蛋白质泛素化蛋白CREB,获得产纤维素酶活性提高的ΔcreB突变株,该突变株不仅能显著提高菌株的滤纸酶活,而且对纤维素内切酶、外切酶、木聚糖酶活均有明显的提高作用,表明CREB很可能也作用于其他 (除CREA之外) 与纤维素酶基因表达调控相关的转录调控因子,并影响到其稳定性。而且,通过图 10的 CreB系统进化树发现斜卧青霉CreB与产黄青霉以及曲霉属的较为相近。在产纤维素酶丝状真菌中,纤维素酶的调控受到多种转录调控因子的作用,如 ACE1[30]、ACE2[30]、CREA[4]、XLNR[22-23]以及其他新发现的转录调控因子 (数据未发表),因此,对转录调控因子稳定性的改造作为1种重要的细胞调控机制对于产酶菌株生产改造具有重要的意义。
由于丝状真菌具有较强的蛋白分泌能力,已成为重要的表达生产内、外源蛋白“细胞工厂”之一。然而由于丝状真菌较强的蛋白酶活性,表达分泌到细胞外的外源蛋白产量仍处于较低水平。CREB的缺失可使突变株分泌胞外蛋白的含量提高2.68倍。细胞外蛋白质含量的增加,有可能由于 creB基因的缺失造成蛋白酶的泛素化,进而造成泛素化的蛋白酶的快速降解,从而间接提高了蛋白分泌到细胞外的含量。因此,深入开展蛋白质的改造对于提高内、外源蛋白的高效表达并提高其稳定性具有重要的作用。
通过同源双交换整合的方式首次在青霉中敲除了编码去蛋白质泛素化的基因creB,该基因的敲除未对菌株的表型产生明显的影响,但可显著提高突变株的纤维素降解利用能力,并且明显增加胞外蛋白质的含量。斜卧青霉作为重要的产纤维素酶工业生产菌株,其基因组序列已测序完成,因此,利用基因工程手段并结合组学的研究方法对菌株的代谢途径进行系统改造已成为构建高效工业生产菌株重要的创造平台。
[1] Gao L, Wang FH, Gao F, et al. Purification and characterization of a novel cellobiohydrolase (PdCel6A) from Penicillium decumbens JU-A10 for bioethanol production. Bioresour Technol, 2011, 102(17): 8339−8342.
[2] Chen S, Xing XH, Huang JJ, et al. Enzyme-assisted extraction of flavonoids from Ginkgo biloba leaves: Improvement effect of flavonol transglycosylation catalyzed by Penicillium decumbens cellulase. Enzyme Microb Technol, 2011, 48(1): 100−105.
[3] Wei XM, Qin YQ, Qu YB. Molecular cloning and characterization of two major endoglucanases from Penicillium decumbens. J Microbiol Biotechnol, 2010, 20(2): 265−270.
[4] Portnoy T, Margeot A, Linke R, et al. The CRE1 carbon catabolite repressor of the fungus Trichoderma reesei: a master regulator of carbon assimilation. BMC Genomics, 2011, 12(1): 269.
[5] Lockington RA, Kelly JM. Carbon catabolite repression in Aspergillus nidulans involves deubiquitination. Mol Microbiol, 2001, 40(6): 1311−1321.
[6] Lockington RA, Kelly JM. The WD40-repeat protein CreC interacts with and stabilizes the deubiquitinating enzyme CreB in vivo in Aspergillus nidulans. Mol Microbiol, 2002, 43(5): 1173−1182.
[7] Boase NA, Kelly JM. A role for creD, a carbon catabolite repression gene from Aspergillus nidulans, in ubiquitination. Mol Microbiol, 2004, 53(3): 929−940.
[8] Nehlin JQ, Carberg M, Ronne H. Control of yeast GAL genes by Mig1p repressor: a transcriptional cascade in the glucose response. EMBO J, 1991, 10: 3373−3377.
[9] Arst HN, Tollervey D, Dowzer CEA, et al. An inversion truncating the creA gene of Aspergillus nidulans results in carbon catabolite derepression. Mol Microbiol, 1990, 4(5): 851−854.
[10] Rogers S, Wells R, Rechsteiner M. Amino acid sequences common to rapidly degraded proteins: the PEST hypothesis. Science, 1986, 234(4774): 364−368.
[11] Denton JA, Kelly JM. Disruption of Trichoderma reesei cre2, encoding an ubiquitin C-terminal hydrolase, results in increased cellulase activity. BMC Biotechnol, 2011, 11(1): 103.
[12] Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. 3rd ed. New York: Cold Spring Harbor Laboratory Press, 2001.
[13] Li ZH. Development of a highly efficient gene targeting system allowing rapid genetic manipulations and functional analysis of the transcriptional regulator CreA in Penicillium decumbens[D]. Jinan: Shandong University, 2010.李忠海. 斜卧青霉高效基因打靶系统的构建与转录调控因子 CreA功能的研究[D]. 济南: 山东大学, 2010.
[14] Sun XY. Studies on the Synthesis Regulation of lignoeellulose-degrading enzymes in Penicillium decumbens[D]. Jinan: Shandong University, 2007.孙宪昀. 斜卧青霉木质纤维素酶系的合成调控研究[D]. 济南: 山东大学, 2007.
[15] Yu JH, Hamari Z, Han KH, et al. Double-joint PCR: a PCR-based molecular tool for gene manipulations in filamentous fungi. Fungal Genet Biol, 2004, 41(11): 973−981.
[16] Kubodera T, Yamashita N, Nishimura A. Pyrithiamine resistance gene (ptrA) of Aspergillus oryzae: cloning, characterization and application as a dominant selectable marker for transformation. Biosci Biotechnol Biochem, 2000, 64(7): 1416−1421.
[17] Honda S, Selker EU. Tools for fungal proteomics: multifunctional Neurospora vectors for gene replacement, protein expression and protein purification. Genetics, 2009, 182(1): 11−23.
[18] Ninomiya Y, Suzuki K, Ishii C, et al. Highly efficient gene replacements in Neurospora strains deficient for nonhomologous end-joining. Proc Natl Acad Sci USA, 2004, 101(33): 12248−12253.
[19] Lara-Rojas F, Sánchez O, Kawasaki L, et al. Aspergillus nidulans transcription factor AtfA interacts with the MAPK SakA to regulate general stress responses, development and spore functions. Mol Microbiol, 2011, 80(2): 436−454.
[20] Carvalho NDSP, Arentshorst M, Kooistra R, et al. Effects of a defective ERAD pathway on growth and heterologous protein production in Aspergillus niger. Appl Microbiol Biotechnol, 2011, 89(2): 357−373.
[21] Li ZH, Du CM, Zhong YH, et al. Development of a highly efficient gene targeting system allowing rapid genetic manipulations in Penicillium decumbens. Appl Microbiol Biotechnol, 2010, 87(3): 1065−1076.
[22] van Peij NNME, Brinkmann J, Vršanská M, et al. β-Xylosidase activity, encoded by xlnD, is essential for complete hydrolysis of xylan by Aspergillus niger but not for induction of the xylanolytic enzyme spectrum. Eur J Biochem, 1997, 245(1): 164−173.
[23] van Peij NNME, Gielkens MMC, De Vries RP, et al. The transcriptional activator XlnR regulates both xylanolytic and endoglucanase gene expression in Aspergillus niger. Appl Environ Microbiol, 1998, 64(10): 3615−3619.
[24] Sun J, Glass NL. Identification of the CRE-1 cellulolytic regulon in Neurospora crassa. PLoS One, 2011, 6(9): e25654.
[25] Portnoy T, Margeot A, Seidl-Seiboth V, et al. Differential regulation of the cellulase transcription factors XYR1, ACE2, and ACE1 in Trichoderma reesei strains producing high and low levels of cellulase. Eukaryot Cell, 2011, 10(2): 262−271.
[26] Andersen MR, Nielsen ML, Nielsen J. Metabolic model integration of the bibliome, genome, metabolome and reactome of Aspergillus niger. Mol Syst Biol, 2008, 4(1): 178.
[27] Furukawa T, Shida Y, Kitagami N, et al. Identification of specific binding sites for XYR1, a transcriptional activator of cellulolytic and xylanolytic genes in Trichoderma reesei. Fungal Genet Biol, 2009, 46(8): 564−574.
[28] Tamayo EN, Villanueva A, Hasper AA, et al. CreA mediates repression of the regulatory gene xlnR which controls the production of xylanolytic enzymes in Aspergillus nidulans. Fungal Genet Biol, 2008, 45(6): 984−993.
[29] Hasper AA, Trindade LM, van der Veen D, et al. Functional analysis of the transcriptional activator XlnR from Aspergillus niger. Microbiology, 2004, 150(5): 1367−1375.
[30] Saloheimo A, Aro N, Ilmén M, et al. Isolation of the ace1 gene encoding a Cys2-His2transcription factor involved in regulation of activity of the cellulase promoter cbh1 of Trichoderma reesei. J Biol Chem, 2000, 275(8): 5817−5825.
猜你喜欢
生物化学与生物物理进展(2022年8期)2022-08-20
传染病信息(2022年2期)2022-07-15
现代临床医学(2022年3期)2022-06-06
汉字汉语研究(2021年2期)2021-08-30
汉字汉语研究(2019年2期)2019-08-27
天津医科大学学报(2019年3期)2019-08-13
新高考·英语进阶(高二高三)(2018年8期)2018-01-15
河北书画研究(2016年3期)2016-04-28
浙江大学学报(农业与生命科学版)(2015年4期)2015-12-15
中国医学科学院学报(2015年5期)2015-03-01
