
7例散发性Creutzfeldt-Jakob病患者的PRNP和APOE基因突变/变异检测及其临床意义
2012-01-26 02:55李骄星孙逊沙盛文利
中风与神经疾病杂志 2012年8期
罗 曼, 李骄星, 孙逊沙, 盛文利
Creutzfeldt-Jakob病简称CJD,其年发病率约为百万分之一,是以快速进展性痴呆为特征,伴有小脑、锥体系和锥体外系受损症状和体征的神经系统变性疾病。本病的发病类型可分为散发性、家族性、医源性和新变异型等4种形式,其中散发性CJD约占所有CJD的85%[1]。CJD是由一种特殊的具有感染性的蛋白质-朊蛋白(prion protein,PrP)所引起,而该蛋白由朊蛋白基因(prion protein gene,PRNP)编码。另外,载脂蛋白E(apolipoprotein E,APOE)基因的ε4等位基因已被广泛认可与阿尔茨海默病的发病有关,鉴于痴呆为CJD的主要临床表现,国外学者对白种人CJD患者进行了APOE基因检测,研究表明ε4等位基因可能也是CJD的危险因子[2]。本研究拟对中国汉族人群散发性CJD患者进行PRNP基因及APOE基因检测,了解其突变/变异对散发性CJD发病及表型的影响。
1 对象与方法
1.1 对象 包括7例散发性 CJD患者,为2010年8月~2011年8月中山大学第一附属医院神经内科患者。男性4例,女性3例。发病年龄为52~76岁,平均62.0±8.2岁。7例患者均进行了详细的内科及神经系统专科检查,并进行了脑电图、MRI、脑脊液14-3-3蛋白等辅助检查。诊断参照WHO提出的 CJD诊断标准[3],排除脑血管病、炎症、肿瘤、外伤、代谢紊乱等疾病引起的痴呆,7例患者均诊断为很可能的(probable)CJD。全部患者无明确家族史,且均为中国汉族人群。
1.2 方法 抽取患者外周静脉血4ml,置EDTA抗凝管中。血样用血液基因组DNA提取试剂盒(北京TIANGEN公司)提取DNA,用核酸蛋白测定仪测定DNA纯度及浓度,二者均合格后保存于-20℃冰箱中备用。参照文献合成PRNP基因开放阅读 框 架 引 物[4](F:5’-CAGAGCAGTCATTATGGCGAACCT-3’,R:5’-AGACCTTCCTCATCCCACTATCAG-3’)及 APOE 基因第四外显子引物[5](F:5’-AACAACTGACCCCGGTGGCG-3’,R:5’-ATGGCGCTGAGGCCGCGCTC-3’),引物由英潍捷基(上海)贸易有限公司合成。以基因组DNA为模板,应用聚合酶链反应(polymerase chain reaction,PCR)扩增基因片段。PCR反应体系为25μl,其中包括:DNA模板1μl,上、下游引物(20pmol/μl)各 0.25μl,10 × PCR buffer 2.5μl,dNTP(2.5mmol)2μl,Taq 酶(5U/μl)0.2μl,灭菌双蒸水 18.8μl。PCR 反应条件为:94℃ 预变性5min,94℃变性 30s,58℃ 或 65℃ 退火 30s,72℃ 延伸1min,共30个循环,最后72℃再延伸10min。PRNP基因退火温度为65℃,APOE基因为58℃。PCR扩增产物经1%琼脂糖凝胶电泳检测,溴化乙锭染色。目的产物送广州赢森生物科技有限公司测序。
2 结果
2.1 PRNP基因测序 发现1例患者(病例1)存在PRNP基因E200K杂合突变(G598A),经反向测序证实。结果(见图1)。病例1临床特点为:女性,58岁发病,亚急性起病,进展迅速。以记忆力减退及失语为首发症状,认知功能障碍进行性加重并逐渐出现小脑症状(头晕、行走不稳)、锥体外系症状(四肢不自主运动)、肌阵挛、缄默。病程2个月时出现意识障碍并逐渐加重。头部MRI示双侧扣带回、双侧丘脑等T1、T2长信号,脑干、小脑未见异常。脑电图示广泛性中波幅慢波,左侧颞区、额区间断性类周期性癫痫样放电,有时可波及到同侧其他导联。送广东省疾控中心检测脑脊液14-3-3蛋白(+)。发现6例患者PRNP基因M129V多态性(A385G)均为129基因型MM,仅1例患者(病例2)为129基因型MV,结果(见图2)。未发现国外报道[6]的可能与CJD致病相关的位点突变(G114V、R148H、D178N、V180I、T183A、H187R、T188K、E196K、V203I、R208H、V210I、E211Q、M232R、P238S)。
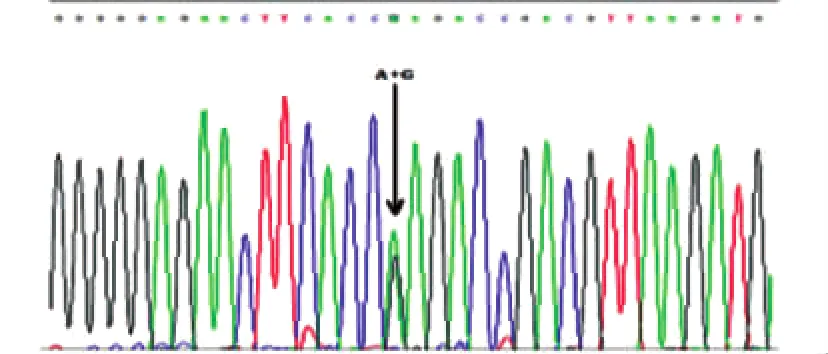
图1 病例1的PRNP基因测序结果示E200K杂合突变(G598A)
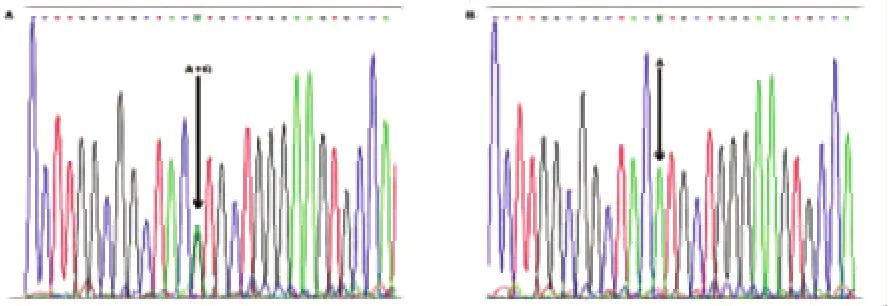
图2 PRNP基因M129V多态性测序结果
2.2 APOE基因测序 发现1例患者(病例3)存在 APOE基因 ε4等位基因,基因型为 E3/4(ε3ε4),其余6例患者都未检测到该多态性,即基因型为E3/3(ε3ε3)。病例3临床特点为:男性,52岁发病,亚急性起病,进展迅速。以小脑症状(头晕、行走不稳)为首发症状,1w后出现进行性加重的认知功能障碍,并逐渐出现锥体外系症状(四肢不自主运动)、肌阵挛、缄默。病程1个月余即出现意识障碍并逐渐加重。头部MRI+DWI示双侧额颞顶皮层DWI不对称信号增强,脑皮层及小脑萎缩。脑电图示中重度异常,未见周期性放电。送广东省疾控中心检测脑脊液14-3-3蛋白(+)。
3 讨论
3.1 PRNP基因致病性突变检测 人类PRNP基因位于20号染色体短臂,共约20kb碱基。它包含2个外显子,第一外显子为非编码外显子,第二外显子含有795bp的开放阅读框架。现有研究表明,散发性CJD的临床病理及分子表型在很大程度上会受到PRNP基因变异的影响[7]。国外已报导的位于PRNP基因的CJD致病性突变包括:G114V、R148H、D178N、V180I、T183A、H187R、T188K、E196K、E200K、V203I、R208H、V210I、E211Q、M232R、P238S 等,本研究对7例散发性CJD患者进行了上述位点的检测,结果发现1例患者(病例1)存在PRNP基因E200K杂合突变(G598A),未发现其他位点突变。
E200K是全球范围内PRNP基因的最常见突变,它是200位密码子由GAG突变为AAG,导致谷氨酸(Glu,E)改变为赖氨酸(Lys,K)。该突变最先在斯洛伐克乡村的CJD群集病例中发现[8],几乎同时,利比亚的一个患病犹太家庭也被检测出了该突变[9]。目前,已有超过10个国家在CJD患者中发现了该突变,该突变的临床表型除了常具有严重的睡眠障碍外,总体来说与散发性CJD相似,而睡眠障碍可能与该突变常伴发显著的丘脑损伤有关。2010年,Gao等[10]报道了我国首例E200K突变的CJD患者,该例患者同样以睡眠障碍为典型临床表现,除此之外与散发性CJD无异。次年,中国疾病预防控制中心[11]对2006年~2010年中国克雅氏病监测网络获得的散发性CJD患者特点进行了总结,在所诊断的284例CJD患者中,仅发现1例E200K突变患者,即2010年Gao等所报道的病例。本研究所发现的该例E200K突变患者也具有与散发性CJD相似的特点,如发病年龄、进展性痴呆、小脑症状、锥体外系症状、肌阵挛、EEG及MRI改变、脑脊液14-3-3蛋白阳性等,这与以往报道相一致[6,10,12],但其睡眠障碍并不明显,我们考虑有以下可能:(1)虽然该患者影像学检查也显示双侧丘脑损害,但可能在早期损害较轻,未显示明显的症状;(2)早期的智能障碍部分掩盖了睡眠障碍症状;(3)迅速出现的意识障碍掩盖了睡眠障碍症状。另外,虽然其家族成员均无CJD患者,但由于其出现了致病性突变,仍应归为家族性CJD,我们将联系其子女进行基因检测并追踪其家族成员的发病情况。
3.2 PRNP基因多态性检测 以往研究显示,PRNP基因M129V是与CJD易感性相关的多态性位点[6]。M129V(A385G)多态性是指 PRNP 基因第129位密码子可以为编码甲硫氨酸(Met,M)的ATG,也可以为编码缬氨酸(Val,V)的 GTG。在正常高加索人群中,37%的人群为 Met/Met(MM),51%为 Met/Val(MV),只 有 12%为 Val/Val(VV)[13]。多个研究均显示,该位点 MM 型与 CJD易感性相关,在散发性CJD患者中,MM型所占的比例可以达到70% ~80%[14,15];在E200K突变所致的家族性 CJD中,MM型也显示与 E200K突变相关[6]。本研究所检测的7例CJD患者中共6例为MM型,所占比例高达85.7%。即使去除诊断为家族性CJD的病例1,剩余的6例患者中MM型所占比例亦达到83.3%,这与国外的报道一致,也与以往针对中国散发性CJD患者的研究结果一致[11]。
3.3 APOE基因多态性检测 人类APOE基因位于19号染色体长臂,由299个氨基酸组成,包含4个外显子和3个内含子。最常见的APOE多态性为ε2/ε3/ε4等位基因多态性,该多态性的分子学基础为氨基酸序列112位半胱氨酸(Cys)与158位精氨酸(Arg)互换。ε2在这两个位置均为Cys,ε4则均为Arg,ε3分别为Cys和Arg。这3种等位基因导致了人群中存在6种不同的基因型,即3种杂合子(E2/3、E3/4、E2/4)和3 种纯合子(E2/2、E3/3、E4/4),其中ε3和E3/3是最常见的等位基因和基因型[16],而 ε4等位基因已被广泛认可与痴呆有关。鉴于痴呆为CJD的主要临床表现,我们对CJD患者进行了APOE基因多态性检测,结果发现仅1例患者为E3/4(ε3ε4)基因型,其余6例均为E3/3基因型。该例E3/4基因型患者为所有患者中发病年龄最小(52岁)、疾病进展最快、症状较重的,有别于其他E3/3基因型患者。国外学者曾对CJD患者进行了APOE基因多态性及脑内β-淀粉样蛋白(Aβ)检测,结果发现ε4等位基因与脑内Aβ沉积相关,故得出ε4等位基因是加速CJD进展的危险因子的结论[2]。这可以帮助解释本研究中E3/4基因型患者发病早、进展快的现象。
本研究结合既往的研究结果表明,PRNP基因E200K突变为CJD致病性突变,129基因型MM多态性与散发性CJD易感性相关。本研究将1例E200K突变的家族性CJD患者误诊为散发性CJD患者,鉴于两者在临床表现的相似性,对CJD患者进行常规PRNP基因检测将有助于两者的鉴别,进而有助于对患病家系成员进行早期诊断。然而,由于本研究病例数较少,尚不能肯定APOE基因ε4等位基因对散发性CJD进展的影响确实存在,我们将在今后的工作中进一步加大样本量。
[1]Mead S,Stumpf MP,Whitfield J,et al.Balancing selection at the prion protein gene consistent with prehistoric kurulike epidemics[J].Science,2003,300(5619):640 -643.
[2]Van Everbroeck B,Croes EA,Pals P,et al.Influence of the prion protein and the apolipoprotein E genotype on the Creutzfeldt-Jakob Disease phenotype[J].Neurosci Lett,2001,313(1 ~2):69 -72.
[3]Zerr I,Kallenberg K,Summers DM,et al.Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease[J].Brain,2009,132(10):2659-2668.
[4]Yu SL,Jin L,Sy MS,et al.Polymorphisms of the PRNP gene in Chinese populations and the identification of a novel insertion mutation[J].Eur J Hum Genet,2004,12(10):867 -870.
[5]Richard P,Thomas G,de Zulueta MP,et al.Common and rare genotypes of human apolipoprotein E determined by specific restriction profiles of polymerase chain reaction-amplified DNA[J].Clin Chem,1994,40(1):24 -29.
[6]Brown K,Mastrianni JA.The Prion Diseases[J].J Geriatr Psychiatry Neurol,2010,23(4):277 - 298.
[7]Imran M,Mahmood S.An overview of human prion diseases[J].Virol J,2011,8(1):559 -567.
[8]Hainfellner JA,Parchi P,Kitamoto T,et al.A novel phenotype in familial Creutzfeldt-Jakob disease:prion protein gene E200K mutation coupled with valine at codon 129 and type 2 protease-resistant prion protein[J].Ann Neurol,1999,45(6):812-816.
[9]Hsiao K,Meiner Z,Kahana E,et al.Mutation of the prion protein in Libyan Jews with Creutzfeldt-Jakob disease[J].N Engl J Med,1991,324(16):1091-1097.
[10]Gao C,Shi Q,Zhou W,et al.The first Chinese case of Creutzfeldt-Jakob disease with mutation of E200K in PRNP[J].Biomed Environ Sci,2010,23(2):158 - 160.
[11]Gao C,Shi Q,Tian C,et al.The epidemiological,clinical,and laboratory features of sporadic Creutzfeldt-Jakob disease patients in China:surveillance data from 2006 to 2010[J].PLoS One,2011,6(8):e24231.
[12]Simon ES,Kahana E,Chapman J,et al.Creutzfeldt-Jakob disease profile in patients homozygous for the PRNP E200K mutation[J].Ann Neurol,2000,47(2):257 -260.
[13]Owen F,Poulter M,Collinge J,et al.Codon 129 changes in the prion protein gene in Caucasians[J].Am J Hum Genet,1990,46(6):1215-1216.
[14]Laplanche JL,Delasnerie-Laupretre N,Brandel JP,et al.Molecular genetics of prion diseases in France.French Research Group on Epidemiology of Human Spongiform Encephalopathies[J].Neurology,1994,44(12):2347 -2351.
[15]Salvatore M,Genuardi M,Petraroli R,et al.Polymorphisms of the prion protein gene in Italian patients with Creutzfeldt-Jakob disease[J].Hum Genet,1994,94(4):375 -379.
[16]Zannis VI,Breslow JL,Sangiacomo TR,et al.Characterization of the major apolipoproteins secreted by two human hepatoma cell lines[J].Biochemistry,1981,20(25):7089 -7096.
猜你喜欢
传染病信息(2022年4期)2022-11-23
世界科学技术-中医药现代化(2022年3期)2022-08-22
川北医学院学报(2022年6期)2022-06-24
昆明医科大学学报(2022年2期)2022-03-29
智慧健康(2021年17期)2021-07-30
昆明医科大学学报(2021年3期)2021-07-22
中国产前诊断杂志(电子版)(2020年1期)2020-05-21
遵义医科大学学报(2020年6期)2020-02-05
中国防痨杂志(2018年3期)2018-03-07
中央民族大学学报(自然科学版)(2015年1期)2015-06-11
