
Small rodent models of hepatitis B and C virus replication and pathogenesis
2012-01-23 00:58MarkFeitelsonAllaArzumanyanMarciaClayton
微生物与感染 2012年2期
Mark A. Feitelson, Alla Arzumanyan, Marcia M. Clayton
1. Department of Biology, College of Science and Technology, Temple University, Philadelphia, PA 19122, USA; 2. Center for Biotechnology, Sbarro Health Research Organization, Department of Biology, Temple University, Philadelphia, PA 19122, USA
1 Introduction
There are more than 350 million people worldwide who are carriers of hepatitis B virus (HBV) and 170 million who are chronically infected with hepatitis C virus (HCV)[1,2]. These people are at high risk for the development of chronic hepatitis, cirrhosis, and hepatocellular carcinoma (HCC). HCC is the fifth most frequent tumor type worldwide[3]. Both cirrhosis and HCC are major causes of mortality within 20-40 years after infection[4]. Although screening has now nearly eliminated these viruses from the blood supply[5], significant transmission is still observed sexually (for HBV)[6]and through intravenous drug abuse (for HBV and HCV)[7,8]. Thus, both viruses remain significant public health problems.
Initially, treatment for chronic infections was limited, with interferon α (IFN-α) yielding a sustained response in about 20% of hepatitis B or hepatitis C patients[9,10]. Nucleoside analog-based therapies were then developed with the introduction of lamivudine for HBV[11], and ribavirin for HCV[12], although less than 50% of patients showed a sustained virological and histopathological response after the end of treatment. Prolonged IFN-α treatment had significant side effects[13], while lamivudine-resistant HBV appeared in up to 20% of patients after one year of treatment[14,15]. The other nucleoside analogs that have been developed, were more potent than lamivudine, and did not readily give rise to drug resistance. However, this may change with their continued use. Combination therapies, which would reduce the incidence of drug resistance, have not been adequately assessed in long-term use. Thus, there is a strong mandate to develop relevant animal models to test new drug candidates and combinations against both of these viruses and their associated liver diseases.
2 Natural hosts for HBV and HCV
Part of the difficulty in developing new therapies stems from a lack in understanding the pathogenesis of chronic liver disease (CLD) associated with these viral infections (Fig.1 and Fig.2). Chimpanzees have been invaluable in studying the transmission and natural history of infection, develop cellular immune responses similar to those in humans acutely infected with HBV, and have been used to conduct vaccine studies[16-18]. In the case of HCV, chimpanzees were central to the discovery of the virus[19,20]. Additional work has revealed the central role of CD4+and CD8+T cells in the pathogenesis of acute and chronic infections[21]. However, their expense and endangered status have limited their availability. In addition, the liver disease in experimentally infected chimpanzees is mild, making it difficult to study pathogenesis. Thus, it has not been practical to use chimpanzees for the preclinical evaluation of new therapeutic approaches on a regular basis, or for the evaluation of combination therapies.
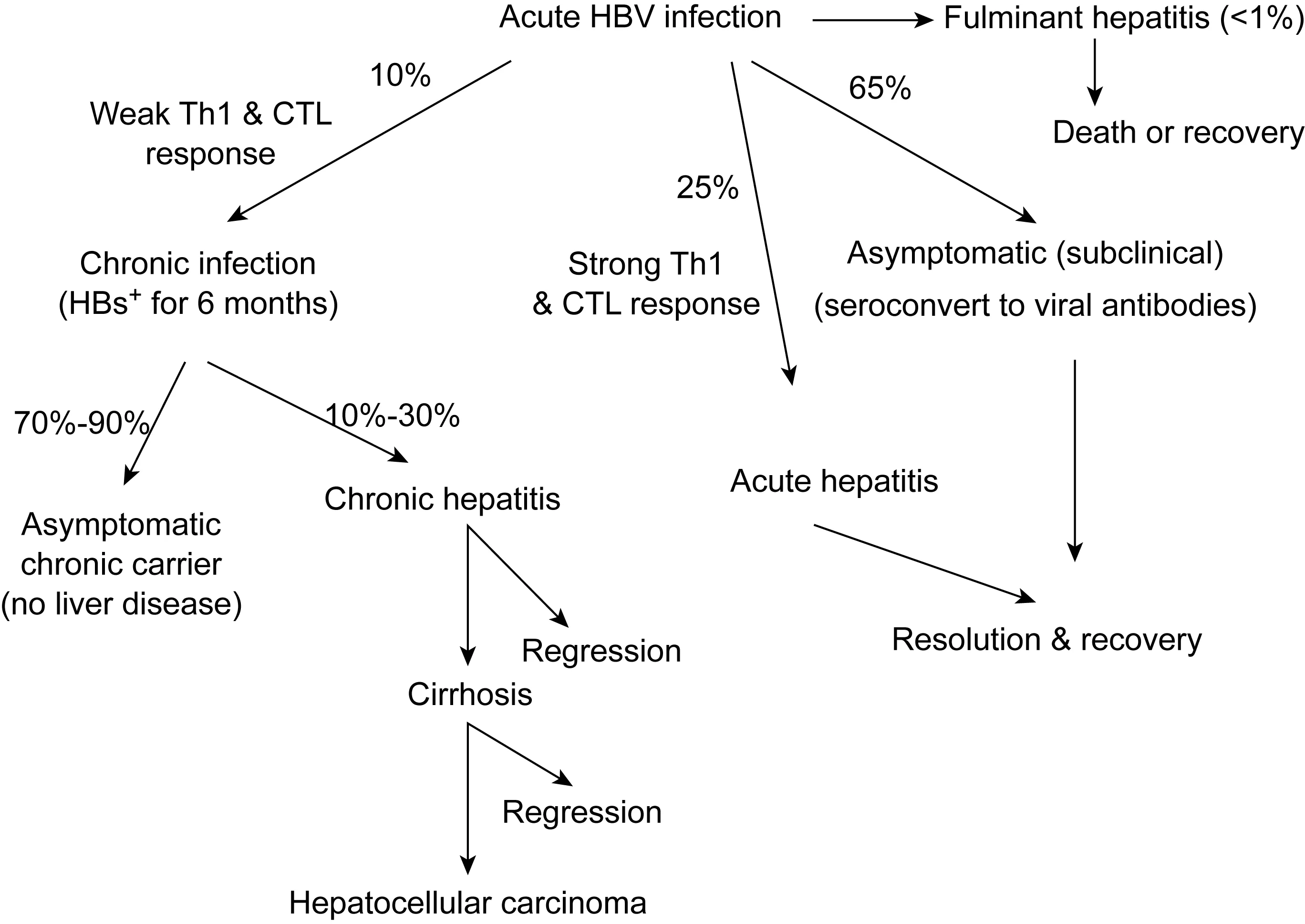
Upon acute exposure to HBV, the majority of adults (about 65%) develop a subclinical infection that is indicted only by the appearance of one or more viral antibodies. Another roughly 25% of infected adults develop a strong Th1 cytokine and cytotoxic T lymphocyte (CTL) response, resulting in a bout of acute hepatitis, followed by resolution and recovery. Rarely, an acute infection rapidly develops into life-threatening fulminant hepatitis. About 10% of acutely infected adults develop a chronic infection, which is characterized by the persistence of HBV surface antigen (HBsAg) and (in some patients) virus in blood. While most chronic carriers remain asymptomatic for years or decades, they are at high risk for the development of chronic hepatitis, cirrhosis, and HCC. Among those that develop liver disease, progression is variable, with some patients developing end-stage liver disease or HCC within a few years, while others undergoing regression of histological lesions in the liver at any stage of CLD.

Acute HCV infection has two major outcomes. Patients developing strong, rapid, multispecific T cell responses often experience either a clinical or subclinical bout of acute hepatitis, followed by resolution, or no liver disease at all. However, the majority of acutely infected patients develop persistent viremia, which is frequently characterized by the appearance of chronic hepatitis. The progression or development of cirrhosis during chronic infection depends upon the characteristics of the immune responses that develop, with more vigorous and sustained immune responses associated with increased liver damage and disease progression. Alternatively, the triggering of weak cell-mediated immune responses after acute infection, or during the course of chronic infection, would favor the development of virus escape mutants that would persist (Modified from ref. 153).
Recently, the tree shrew (Tupaiabelangeri) has been shown to be susceptible to HBV and HCV infections[22-24]. Serial transmission has been achieved using HBV-infected animals, and infection has been prevented by immunization of susceptible animals with the HBV vaccine. Chronically infected animals developed hepatitis, cirrhosis and a low incidence of HCC[25]. For HCV, infection occurred in animals that were immunosuppressed by irradiation prior to infection. This resulted in viremia that was transient or intermittent in about a third of the animals, and for a more extended time in half the animals[22]. The findings that primary tupaia hepatocytes (PTHs) could be infectedinvitrowith serum derived from chronic HCV-infected patients[26,27], and that this system was used to functionally characterize the host-encoded receptors for HCV[28], have further established the tree shrew as a small animal model of HCV which could be exploited in the development of new therapeutics.
3 Hepadnaviruses
Hepadnaviruses[29]include HBV and several related agents that naturally infect selected hosts in the wild. The three best studied are the ground squirrel hepatitis virus (GSHV)[30], the woodchuck hepatitis virus (WHV)[31], and the duck hepatitis B virus (DHBV)[32]. Infected ducks have been very useful in elucidating the replication scheme of hepadnaviruses[33], and primary duck hepatocytes are readily infected with DHBV[34], making them useful for the evaluation of drugs that inhibit virus replication in a fully permissiveinvitrosystem[35,36]. Infected ducks and woodchucks have also been used for preclinical antiviral drug development[37,38]. Infected woodchucks have also been used to elucidate the natural history of infection[39,40], which is similar to that in chronic human infections (Fig.1), and the molecular mechanisms of HCC (Fourel,etal., 1994; Hsu,etal., 1988; Popper,etal., 1987; Snyder,etal., 1982)[41-44], which differs from that in human infections. For example, WHV often integrates at or near the N-mycgene during chronic infection, promotingmycexpression[45], while in chronic human infections, integration has rarely been found in or around themycgene. In addition, neither infected ducks nor woodchucks develop cirrhosis, nor do infected ducks develop HCC. Since the pathogenesis of these infections is immune-mediated, immunological reagents that might have therapeutic efficacy against WHV or DHBV infections would probably not be useful against HBV, and that for human trials, HBV-specific reagents would have to be created and tested. Hence, despite the wealth of knowledge gained from studying hepadnaviruses in their natural hosts, the types of experiments that could be devised to elucidate the mechanisms of pathogenesis, have been limited.
4 Hepaciviruses
The family Flaviviridae contains three genera. Hepaciviruses contain only HCV. Flaviviruses contain insect-borne agents, including yellow fever and Dengue viruses. Pestiviruses include bovine viral diarrhea virus, the latter of which has been used as a surrogate for some aspects of HCV biology[46]. HCV is related to these genera in terms of genome structure, the production of a polyprotein, the structure and function of these polypeptides, and the mode of replication[47,48]. Unlike HBV, there have been no reports of closely related HCV-like viruses that naturally infect wild animals or that can be used to experimentally infect laboratory animals. This restricts the available animal model systems that could be used to understand pathogenesis, and to evaluate putative antiviral compounds or other novel therapeutic approaches. As with HBV, the chimpanzee is susceptible to HCV infection. However, the pathogenesis of chronic infection in chimpanzees is mild and rarely progresses to HCC[49].
5 Mechanisms in the pathogenesis of CLD: cytopathic effects and/or immune-mediated disease?
Direct cytopathic effects (CPEs) occur when virus infection is toxic to the infected cell, resulting in cellular damage, and sometimes cell death. With the exception of the CPE observed in cell lines or transgenic mice that constitutively overexpress HBV antigens[50-52], there is little evidence that the pathogenesis of HBV infection is mediated by CPE. For example, HBV carriers and transgenic mice with sustained and high levels of virus replication have no liver disease[53,54]. Further, tissue culture systems replicating HBV do not develop CPE[55,56]. However, in one transgenic mouse model highly overexpressing HBV surface antigen (HBsAg), intrahepatic accumulation resulted in massive hepatocellular necrosis, followed by extensive regeneration, and then by the appearance of HCC[51,57]. Although this model is not relevant to the pathogenesis of HCC in man, in which there is not even a correlation between HBsAg expression and regions of hepatitis in the liver[58], it demonstrates that persistent and strong inflammation is an important risk factor for tumor development, as noted in earlier work[59].
There is now strong evidence that the pathogenesis of HBV infection is immune-mediated. For example, liver and peripheral blood lymphocytes demonstrate proliferative and cytotoxic activities against HBV antigens[60-62]. HBV-infected patients under immunosuppression often have widespread virus gene expression in the liver and high levels of virus in the serum without liver disease, but these patients often develop severe liver disease once immunosuppressive therapy is terminated and antiviral immune responses reassert themselves[63]. Further, HBsAg or HBV transgenic mice adoptively transferred with virus-specific cytotoxic T lymphocytes (CTLs) develop either acute or fulminant hepatitis[57,64], while HBV transgenic severe combined immunodeficient (SCID) mice adoptively transferred with normal and syngeneic splenocytes develop either acute or chronic hepatitis[65]. Moreover, strong and rapid cell-mediated immune responses to multiple HBV antigens is characteristic of acute and resolving infections, while weak immune responses to few HBV antigens is characteristic of infections that become chronic[58,60,66]. Further studies showed that adoptively transferred CTLs strongly inhibit HBV DNA replication by direct cytotoxicity[67], and by noncytolytic mechanisms via release of cytokines[68-70], which reduce the steady-state levels of viral RNAs in HBV-replicating hepatocytes[71]. Adoptive transfer of HBV-specific CTLs also promotes inflammation by recruiting polymorphonuclear leukocytes to the site of liver cell damage[72]. Independent work showed that back-crossing immunodeficient (RAG1 or TCR knockout) mice with HBsAg transgenic mice, followed by adoptive transfer of splenocyte subpopulations, yielded a model of acute hepatitis B. Further analysis showed that a subset of nonclassical natural killer T (NKT) cells induced acute hepatitis[73]. Hence, the pathogenesis of HBV is immune-mediated, although the virus antigens and corresponding immune responses that are responsible for acutevs. CLD have not been clearly identified.
Flaviviruses tend to mediate pathology by CPE[47], although the case is not as clear with HCV. The finding of severe cholestatic hepatitis in a subset of HCV-infected liver transplant patients[74], and that HCV-associated CLD is more severe in human immunodeficiency virus (HIV)-infected patients compared to those without HIV infection[75], suggest CPE, since these patients are immunodeficient. The HCV-JFH1 strain, which was derived from a Japanese patient with fulminant hepatitis, replicates to a high levelinvitroand demonstrates CPE[76]. In addition, HCV turned on genes that mediate apoptosis in liver transplant patients who experienced re-infection and rapidly developing fibrosis[77]. The development of lymphoid aggregates and follicles in the liver, of bile duct damage, of activated sinusoidal inflammatory cells[78], and of T helper (Th) cells and CTLs in CLD[79], suggests that these lesions were mediated by immune responses against virus-infected hepatocytes. However, the lack of liver pathology in acutely infected chimpanzees during the incubation period of infection, and the persistence of virus in chimpanzees and patients in the absence of liver disease[80], suggest that HCV may not always be directly cytopathic. Thus, both mechanism may contribute to pathogenesis (Fig.2)[81,82]. These features are not only important for the design of relevant animal models, but also for the selection of single and multiple therapeutic approaches that are likely to target the underlying causes of CLD.
6 Mouse and rat models of HBV
Laboratory animals, like mice and rats, are not susceptible to HBV infection. Thus, the development of transgenic mouse technology, permitted the construction of mice that expressed the inserted sequences, so that the function of transgene expression could be studiedinvivo[83]. Accordingly, by 1985, two research groups created transgenic mice that produced HBsAg particles[84-86]that were indistinguishable from those found in human infections. These mice were tolerant to HBsAg, so that despite high levels of circulating antigen, no liver disease was detected, as with the human asymptomatic carrier. Other lines of transgenic mice were made and shown to support HBV replication, but in all cases, no liver disease was observed[53,87,88]. However, overexpression of HBsAg containing preS sequences resulted in their retention and accumulation in the liver of transgenic mice[89]. Age-related accumulation resulted in hepatotoxicity, chronic liver injury, inflammation, regenerative hyperplasia, aneuploidy and finally HCC[50,51]. Although there is no evidence that HBsAg causes toxic liver injury in humans, these studies highlighted the importance of chronic liver cell injury and hepatocellular regeneration to the pathogenesis of HCC. In this way, these mice provide an explanation for the epidemiologic findings that the most important risk factors for HCC were the HBsAg carrier state and progressive CLD (Fig.1)[59].
HBV X antigen (HBxAg) was also shown to play a pivotal role in tumor development. For example, transgenic mice with sustained high levels of X protein in the liver developed HCC in the absence of CLD[90-92]. No HCC was seen in HBx transgenic mice with low or undetectable HBxAg[93,94]. Mechanistic studies showed that HBxAg bound to and inactivated the tumor suppressor, p53[92], which contributes to stepwise carcinogenesis. In addition, HBxAg stimulates the production of transforming growth factor β1 (TGF-β1)[95], which may trigger increased hepatocellular apoptosis and promote fibrogenesis. HBxAg also activates pathways that shift TGF-β1 signaling from those that negatively regulate hepatocellular growth to those that stimulate growth[96,97]. In another line of transgenic mice, HBxAg is absent at birth, but increased with age. These mice were not tolerant to HBxAg, and developed hepatitis, steatosis, dysplasia and finally HCC by 10 months of age[98,99]. These pathological sequence of events, combined with increased HBxAg staining in the liver, were also observed in human infections[100,101]. HBxAg also contributed to elevated reactive oxygen species (ROS) via its association with mitochondria, which is observed in these transgenic mice[102]and in human infections. Elevated ROS, in turn, may contribute to the development of steatosis. Thus, HBxAg contributes to hepatocarcinogenesis in the presence or absence of CLD.
The findings that high and persistent levels of HBsAg in human carriers and transgenic mice occur in the absence of CLD[54], that immunosuppression ameliorates CLD[103], and that HBV replicates in cultured cells without CPE[55,56], suggest that liver cell damage may be immune-mediated[60]. This was supported by the studies cited above, and by evidence showing that HBsAg-specific CTLs adoptively transferred into HBsAg transgenic mice resulted in acute hepatitis. Liver cell injury was confirmed histologically, was major histocompatibility complex (MHC) class I-restricted[64], and was dependent upon CTL-produced interferon γ (IFN-γ). Following adoptive transfer, there was an increase in apoptosis among scattered hepatocytes, followed by the recruitment of antigen nonspecific inflammatory cells, and then the development of necroinflammatory foci that extended beyond the region where CTLs were present[66,72]. Among mice that secreted HBsAg into blood, the disease was transient, nonfatal and destroyed no more than 5% of the hepatocytes. However, when adoptive transfer was performed in transgenic mice that retained HBsAg in the liver, CTL-produced IFN-γ activated intrahepatic macrophages, resulting in fulminant hepatitis and the death of many animals[57].
Immune-mediated acute hepatitis was also independently observed in rats transfected with a replication-competent clone of HBV DNA. When this was conducted in normal rats, HBV DNA appeared in serum within a few days, followed by clearance of the virus, and then by a transient elevation of alanine transaminase (ALT) and histopathological evidence of acute hepatitis. When the same experiment was conducted in T cell-deficient nude rats, no clearance of virus or development of liver disease was observed, suggesting that T lymphocytes play a central role in liver cell injury and the clearance of HBV[104].
While this work established models of acute and fulminant hepatitis, the major clinical problem resides among carriers with CLD (Fig.1). To overcome tolerance, HBsAg transgenic mice were irradiated and thymectomized prior to adoptive transfer. The latter accelerated hepatocellular turnover and the appearance of HCC[105], suggesting that even tumor development has an immune-mediated component. However, only a small percentage of HBsAg carriers develop CLD and HCC. Further, HBsAg is not overexpressed in chronically infected human livers to the levels observed in the HBsAg trangenic mice[106]. Thus, immune-mediated CLD contributes to the development of HCC.
In addition to CTLs, clearance of virus gene expression and replication may also be accomplished by noncytolytic mechanisms involving the production of cytokines (Fig.1). For example, HBsAg-specific CTL clones stimulatedinvitrowith plate-bound anti-CD3 monoclonal antibodies produced IFN-γ, tumor necrosis factor α (TNF-α), and to a lesser extent TNF-β mRNA[57,68]. These cytokine mRNAs correlated with the development of acute hepatitis following adoptive transfer. Interestingly, the administration of monoclonal antibodies against IFN-γ or TNF-α prior to adoptive transfer, largely prevented the CTL-mediated reduction in HBsAg expression, suggesting these cytokines inhibited virus gene expression. These cytokines also inhibited virus replication in HBV transgenic mice[67]. Virus clearance has also been observed without extensive hepatocellular necrosis in WHV-infected animals, even though the great majority of hepatocytes in the woodchuck liver become infected[107]. These data imply that there are not enough virus specific-CTL precursors to mediate direct cytotoxicity with every infected hepatocyte and that cytokines are needed to amplify antigen-specific responses. Among HBsAg transgenic mice, treatment with TNF-α[108]or interleukin 2 (IL-2)[70,109]resulted in reduced steady-state levels of most viral mRNAs among infected hepatocytes[110]. Moreover, administration of IL-12, which induces T and NK cells to produce IFN-γ, inhibits virus replication and gene expression in HBV transgenic mice[111]. When the latter mice were infected with lymphocytic choriomeningitis virus (LCMV), the cytokines triggered by LCMV-activated macrophages in the liver (e.g., TNF-α and IFN-α/β) effectively suppressed HBV replication[69]. These events may also partially explain the suppression of HBV replication in many HCV co-infected patients[112].
The development of CLD in HBV carriers (Fig.1) is a major target for therapeutics, and yet none of the HBV transgenic models available develop CLD, since they are tolerant to the products of the transgene. In contrast, people who are acutely infected with HBV (or HCV) are immunologically naïve to the virus. To deal with the issue of tolerance, transgenic mice supporting HBV replication were made using SCID hosts that lacked mature T and B cells. Since T and B cells account for the bulk of specific antiviral immunity, these transgenic mice were not tolerant to HBV. Thus, a single adoptive transfer of 107unprimed and syngeneic splenocytes resulted in CLD, while a similar transfer of 5×107cells resulted in acute and resolving hepatitis. Hepatitis was accompanied by mononuclear infiltrates resembling acute and chronic hepatitis in man, and with the clearance of virus gene expression and replicative forms from the liver, as well as clearance of HBsAg and virus DNA from the blood[65]. Additional work with this model has shown that acute and resolving hepatitis is associated with a strong Th1 response and a high CD8∶CD4 ratio, while CLD is associated with a predominantly Th2 response and a low CD8∶CD4 ratio (Feitelson,etal., unpublished data).
HBV transgenic mice have also been used to explore the antiviral efficacy of RNAi[113-115]. In addition, HBV transgenic mice have been used to characterize nucleoside analogs[116]and to develop new antiviral compounds[117]. Further, HBV transgenic mice have been used for the preclinical development of a therapeutic vaccine based upon HBsAg-primed dendritic cells[118]. Thus, HBV transgenic mice provide opportunities for the characterization of next generation therapeutics against the virus.
7 Transgenic mice for HCV
The narrow host range and lack of suitable tissue culture systems for HCV have provided significant barriers to studying the basic biology of host-virus interactions (Fig.2), including pathogenesis, and for testing putative antiviral compounds. Therefore, several groups have developed transgenic models of HCV gene expression and replication. In one study, transgenic mice were made using the HCV E1 and E2 genes[119]. These animals expressed E1/E2 in many organs, including the liver, but did not develop liver disease in mice up to 16 months of age, suggesting that E1/E2 was not cytopathic. However, these mice developed sialadenitis resembling Sjogrens syndrome[120], which is an autoimmune disease affecting the salivary glands, and is associated with HCV infection in man[121]. Independent work showed that HCV core plus E2 transgenic mice did not develop CLD or HCC[122], suggesting that core was not directly cytopathic. Other transgenic mice expressing E1, E2 plus core also had normal liver histopathology[123,124]. Immunization of these mice with a DNA vaccine making HCV core and IL-2 induced significant CD4+and CD8+T cell responses, suggesting that tolerance could be broken[125]. Tolerance was also circumvented with the construction of transgenic mice that conditionally expressing core, E1 and E2 using the Cre/loxP system, but upon expression of these proteins, no liver pathology developed[126]. In contrast, a different lineage of transgenic mice making HCV envelope and core developed focal inflammation, hepatocellular necrosis and degeneration, as well as altered foci with mitotic figures by 10 months of age, compared to nontransgenic controls, suggesting CPE[124]. However, independent production of core transgenic mice resulted in the development of steatosis in mice older than 3 months[127]. When these mice were held beyond 16 months of age, they developed adenomas and then poorly differentiated HCC[128]. HCV core was detected in most adenomas and HCCs in 25%-30% of the animals from two separate lines, suggesting that it is directly oncogenic. Importantly, chronic inflammation, followed by steatosis, precedes the appearance of HCC in human HCV infections. Although the differences between these and other core transgenic mice are not clear, the mice that develop liver pathology express sustained and high levels of core. Although core is found in the cytoplasm of infected cells replicating HCV, its location in the nuclei of hepatocytes in transgenic mice that develop tumors suggests that HCV core may act as a transcriptional regulator that promotes tumor development[129,130]. However, the intrahepatic levels of virus gene expression in these transgenic models are often far higher than in the infected human liver, which raises questions as to how relevant the transgenic systems are to understanding the pathogenesis of HCC in man. Another group developed transgenic mice that express low levels of HCV polyprotein under the transcriptional control of the albumin promoter[131]. These mice develop steatosis, which may result from impairment of mitochondrial fatty acid oxidation[128], accumulation of free radicals, and/or oxidative stress.
Additional transgenic lines were made that carried the full-length HCV cDNA and supported HCV replication[132]. HCV gene expression was generally low, and no consistent histological changes were observed in the liver, suggesting that HCV replication is not directly cytopathicinvivo. Thus, the question still remains as to whether the likely immune-mediated pathogenesis of HCV, as suggested by the clinical literature (Fig.2), can be reproduced in a transgenic model. To probe this question, conditional expression of the HCV envelope and core genes were carried out using the Cre/loxP system[126]. When transgene expression was turned on, expression of envelope and core proteins was detected in the liver within a week, followed by a transient influx of T cells and a spike in transaminases. When transgene induction was repeated in T cell-depleted mice, there was normal histology and baseline transaminase level, suggesting the inflammatory response and hepatocellular damage were T cell-mediated. By day 14 after transgene activation, core was cleared from serum and replaced with corresponding antibodies[126]. Independent work, in which transgenic mice carrying full-length HCV were crossed with HBxAg transgenic mice, showed an accelerated appearance and progression of CLD to HCC[133], suggesting that HBV and HCV co-infections elevated the risk for CLD and HCC, which may be relevant to understanding these virus interactions in human co-infections.
8 Mouse-human liver chimeric models
The early steps in HBV and HCV replication, and the mechanisms involved in virus elimination, cannot be properly studied in transgenic mice because they are not fully permissive for these viruses. Thus, mouse models were devised which consist of transplanting or injecting human hepatocytes so that they could survive for a prolonged period, be infected, and then studiedinvivo. The first model capable of supporting human hepatocytes long-term was the human trimera mouse[134]. In this model, Balb/c mice were lethally irradiated, reconstituted with SCID mouse bone marrow cells, and then transplanted with HBV-infected human liver biopsies under the kidney capsule. Viremia developed for about 20 days, thereby permitting short-term assessment of antiviral drugs. To extend the life-time of transplanted hepatocytes, human liver cells suspended in matrigel were transplanted under the kidney capsule of nonobese diabetic (NOD)/SCID mice. Treatment of these mice with anti-c-Met (where Met is the hepatocyte growth factor receptor) improved hepatocyte survival, so that upon infection with HBV, viremia was observed for up to 5 months[135]. In another approach, immortalized human hepatocytes stably transfected with HBV were injected into the spleens of RAG2-deficient mice. High titers of virus were observed for at least 5 months[136], suggesting that long-term virus replication could be achievedinvivo.
Although useful, only a modest number of human hepatocytes can seed intact mouse livers, and those that do showed little proliferation. To address this, mice were genetically engineered for the hepatocyte-targeted expression of the albumin-urokinase plasminogen activator (uPA) transgene. This resulted in the death of transgene-carrying hepatocytes, which provided a growth advantage for transplanted cells[137]. When uPA transgenic mice were crossed with immunodeficient RAG2 mice (which lack mature T and B cells), intrasplenic injection of primary woodchuck hepatocytes resulted in a repopulation of almost the entire mouse liver. These mice were then productively infected with WHV[138]. Human hepatocytes were also successfully transplanted into uPA/RAG2 mice, and following integration into the mouse liver, such hepatocytes were susceptible to both HBV and HCV[139,140]. Refinement of this model in uPA/SCID mice resulted in the repopulation of almost the entire mouse liver, and following HBV infection, high titers of virus (up to 1010copies/ml) were observed for up to 5 months after the injection of HBV-positive human serum[141,142]. This approach has been used to characterize a putative inhibitor of the HCV RNA-dependent RNA polymerase, HCV-796[143], to evaluate the pharmacology of the specific NS3-4A protease inhibitor, telaprevir[144], and to characterize other antiviral approaches[145]. Until recently, the high mortality of these transgenic mice, and the difficulties of getting a constant sources of primary human hepatocytes, has limited their use. However, with the increasing availability of commercially available human hepatocytes, and with conditions optimized for engraftment and infection[146], it is likely that this model will continue to be used.
9 Xenograph models
Immunodeficient mice have also been used for the transplantation of human cells supporting HBV and HCV replication or a reporter of virus replication in order to evaluate putative antiviral compoundsinvivo. For example, AD38 cells, in which HBV replication is under the control of the tetracycline repressor[147], have been used to grow subcutaneous tumors in nude mice. As the tumors grow, these mice become viremic, and have been used for the development of combination therapies against HBV[148]. Likewise, Huh7 cells replicating HCV injected into beige/SCID mice resulted in viremia, which was inhibited by treatment with human IFN-α and with the HCV protease inhibitor, BILN-2061[149]. An independent model in SCID mice using Huh7 cells carrying an HCV replicon promoting luciferase (reporter) activity was also sensitive to IFN-α and BILN[150]. Alternatively, when HepG2 cells stably replicating an infectious clone of HCV was injected subcutaneously into SCID mice, viremia was observed, and this was also sensitive to treatment with IFN-α[151]. Another variation on this theme was published in which fetal rats were tolerizedinuterowith Huh7 cells, and after birth, were then transplanted with Huh7 cells, and finally infected with HCV. Infected Huh7 cells were found in the liver, where they actively replicated and secreted HCV into the blood. Mild hepatitis was observed, suggesting that both replication and CLD could be studied[152].
10 Conclusions
There is considerable evidence that the pathogenesis of HBV and HCV infections is immune-mediated. The challenge has been developed easily manipulated animal models to study these human virus infections, and in particular, to understand the pathogensis of disease as well as development of HCC. While progress has been made in this direction with the development of several transgenic mouse models, additional models need to be generated that encompass a broader range of host-virus interactions, especially with regard to the pathogenesis of CLD. If this is accomplished, then such models will be very useful for understanding the mechanisms of pathogenesis, and for the evaluation of new therapeutic approaches aimed at both the virus and associated CLDs.
[1] Delwaide J, Gérard C. Evidence-based medicine treatment of chronic hepatitis C. Liege Study Group on Viral Hepatitis [J]. Rev Med Liege, 2000, 55 (5): 337-340.
[2] Wild CP, Hall AJ. Primary prevention of hepatocellular carcinoma in developing countries [J]. Mutat Res, 2000, 462 (2-3): 381-393.
[3] Parkin DM, Stjernsward J, Muir CS. Estimates of the worldwide frequency of twelve major cancers [J]. Bull World Health Organ, 1984, 62 (2):163-182.
[4] Alter MJ. Epidemiology and disease burden of hepatitis B and C [J]. Antiviral Ther, 1996, 1(Suppl 1): 9-14.
[5] Gerlich WH, Caspari G. Hepatitis viruses and the safety of blood donations [J]. J Viral Hepat, 1999, 6(Suppl 1): 6-15.
[6] Mast EE, Williams IT, Alter MJ, Margolis HS. Hepatitis B vaccination of adolescent and adult high-risk groups in the United States [J]. Vaccine, 1998, 16(Suppl): S27-S29.
[7] Maddrey WC. Hepatitis B: an important public health issue [J]. J Med Virol, 2000, 61 (3):362-366.
[8] Smyth R, Keenan E, Dorman A, O’Connor J. Hepatitis C infection among injecting drug users attending the National Drug Treatment Centre [J]. Ir J Med Sci, 1995, 164 (4):267-268.
[9] Lindsay KL. Therapy of hepatitis C: overview [J]. Hepatology, 1997, 26(3 Suppl 1): 71S-77S.
[10] Torresi J, Locarnini S. Antiviral chemotherapy for the treatment of hepatitis B virus infections [J]. Gastroenterology, 2000, 118(2 Suppl 1):S83-S103.
[11] Dusheiko G. Lamivudine therapy for hepatitis B infection [J]. Scand J Gastroenterol, 1999, 34(Suppl 230): 76-81.
[12] Reichard O, Schvarcz R, Weiland O. Therapy of hepatitis C: alpha interferon and ribavirin [J]. Hepatology, 1997, 26(3 Suppl 1): 108S-111S.
[13] Hoofnagle JH, Lau D. Chronic viral hepatitis—benefits of current therapies [J]. N Engl J Med, 1996, 334 (22):1470-1471.
[14] Honkoop P, Niesters HG, de Man RA, Osterhaus AD, Schalm SW. Lamivudine resistance in immunocompetent chronic hepatitis B. Incidence and patterns [J]. J Hepatol, 1997, 26 (6): 1393-1395.
[15] Tipples GA, Ma MM, Fischer KP, Bain VG, Kneteman NM, Tyrrell DL. Mutation in HBV RNA-dependent DNA polymerase confers resistance to lamivudine in vivo [J]. Hepatology,1996, 24 (3):714-717.
[16] Bertoni R, Sette A, Sidney J, Guidotti LG, Shapiro M, Purcell R, Chisari FV. Human class I supertypes and CTL repertoires extend to chimpanzees [J]. J Immunol, 1998, 161(8):4447-4455.
[17] Prince AM, Brotman B. Biological and immunolgical aspects of hepatitis C virus infection in chimpanzees [J]. Curr Stud Hematol Blood Transfus, 1998, 62:250-265.
[18] Tabor E, Purcell RH, Gerety RJ. Primate animal models and titered inocula for the study of human hepatitis A, hepatitis B, and non-A, non-B hepatitis [J]. J Med Primatol, 1983, 12 (6):305-318.
[19] Alter HJ, Purcell RH, Holland PV, Popper H. Transmissible agent in non-A, non-B hepatitis [J]. Lancet, 1978, 1 (8062):459-463.
[20] Bradley DW, Cook EH, Maynard JE, McCaustland KA, Ebert JW, Dolana GH, Petzel RA, Kantor RJ, Heilbrunn A, Fields HA, Murphy BL. Experimental infection of chimpanzees with anti-hemophilic (factor VIII) materials: recovery of virus-like particles associated with non-A, non-B hepatitis [J]. J Med Virol, 1979, 3(4):253-269.
[21] Shoukry NH, Cawthon AG, Walker CM. Cell-mediated immunity and the outcome of hepatitis C virus infection [J]. Annu Rev Microbiol, 2004, 58: 391-424.
[22] Xie ZC, Riezu-Boj JI, Lasarte JJ, Guillen J, Su JH, Civeira MP, Prieto J. Transmission of hepatitis C virus infection to tree shrews [J]. Virology, 1998,244(2):513-520.
[23] Yan RQ, Su JJ, Huang DR, Gan YC, Yang C, Huang GH. Human hepatitis B virus and hepatocellular carcinoma. I. Experimental infection of tree shrews with hepatitis B virus [J]. J Cancer Res Clin Oncol, 1996, 122 (5):283-288.
[24] Yan RQ, Su JJ, Huang DR, Gan YC, Yang C, Huang GH. Human hepatitis B virus and hepatocellular carcinoma. II. Experimental induction of hepatocellular carcinoma in tree shrews exposed to hepatitis B virus and aflatoxin B1 [J]. J Cancer Res Clin Oncol, 1996, 122(5):289-295.
[25] Amako Y, Tsukiyama-Kohara K, Katsume A, Hirata Y, Sekiguchi S, Tobita Y, Hayashi Y, Hishima T, Funata N, Yonekawa H, Kohara M. Pathogenesis of hepatitis C virus infection in Tupaia belangeri [J]. J Virol, 2010, 84(1): 303-311.
[26] Xu X, Chen H, Cao X, Ben K. Efficient infection of tree shrew (Tupaia belangeri) with hepatitis C virus grown in cell culture or from patient plasma [J]. J Gen Virol, 2007, 88 (Pt 9):2504-2512.
[27] Jia ZS, Du DW, Lei YF, Wei X, Yin W, Ma L, Lian JQ, Wang PZ, Li D, Zhou YX. Scavenger receptor class B type I mediates cell entry of hepatitis C virus [J]. J Int Med Res, 2008, 36(6): 1319-1325.
[28] Tong Y, Zhu Y, Xia X, Liu Y, Feng Y, Hua X, Chen Z, Ding H, Gao L, Wang Y, Feitelson MA, Zhao P, Qi ZT. Tupaia CD81, SR-BI, claudin-1 and occludin support hepatitis C virus infection [J]. J Virol, 2011, 85(6): 2793-2802.
[29] Robinson WS, Marion P, Feitelson MA, Siddiqui A. The hepadnavirus group: Hepatitis B and related viruses [M]. In: Szmuness W, Alter HJ, Maynard JE. eds. Viral Hepatitis: 1981 International Symposium. Philadelphia: Franklin Institute Press, 1982: 57-68.
[30] Marion PL, Oshiro LS, Regnery DC, Scullard GH, Robinson WS. A virus in Beechey ground squirrels that is related to hepatitis B virus of humans [J]. Proc Natl Acad Sci USA, 1980, 77 (5):2941-2945.
[31] Summers J, Smolec JM, Snyder R. A virus similar to human hepatitis B virus associated with hepatitis and hepatoma in woodchucks [J]. Proc Natl Acad Sci USA, 1978, 75 (9):4533-4537.
[32] Mason WS, Seal G, Summers J. Virus of Pekin ducks with structural and biological relatedness to human hepatitis B virus [J]. J Virol, 1980, 36 (3): 829-836.
[33] Summers J, Mason WS. Replication of the genome of a hepatitis B-like virus by reverse transcription of an RNA intermediate [J]. Cell, 1982, 29(2): 403-415.
[34] Pugh JC, Summers JW. Infection and uptake of duck hepatitis B virus by duck hepatocytes maintained in the presence of dimethyl sulfoxide [J]. Virology, 1989, 172 (2):564-572.
[35] Colledge D, Locarnini S, Shaw T. Synergistic inhibition of hepadnaviral replication by lamivudine in combination with penciclovir in vitro [J]. Hepatology, 1997, 26 (1): 216-225.
[36] Schultz U, Chisari FV. Recombinant duck interferon gamma inhibits duck hepatitis B virus replication in primary hepatocytes [J]. J Virol, 1999, 73 (4):3162-3168.
[37] Hurwitz SJ, Tennant BC, Korba BE, Gerin JL, Schinazi RF. Pharmacodynamics of (-)-beta-2’,3’-dideoxy-3’-thiacytidine in chronically virus-infected woodchucks compared to its pharmacodynamics in humans [J]. Antimicrob Agents Chemother, 1998, 42 (11):2804-2809.
[38] Mason WS, Cullen J, Saputelli J, Wu TT, Liu C, London WT, Lustbader E, Schaffer P, O’Connell AP, Fourel I, Aldrich C, Jilbert AR. Characterization of the antiviral effects of 2’-carbodeoxyguanosine in ducks chronically infected with duck hepatitis B virus [J]. Hepatology, 1994, 19(2):398-411.
[39] Korba BE, Cote PJ, Wells FV, Baldwin B, Popper H, Purcell RH, Tennant BC, Gerin JL. Natural history of woodchuck hepatitis virus infection during the course of experimental viral infection: molecular virologic features of the liver and lymphoid tissues [J]. J Virol, 1989, 63(3):1360-1370.
[40] Korba BE, Brown TL, Wells FV, Baldwin B, Cote PJ, Steinberg H, Tennant BC, Gerin JL. Natural history of experimental woodchuck hepatitis virus infection: molecular virologic features of the pancreas, kidney, ovary and testis [J]. J Virol, 1990, 64(9):4499-4506.
[41] Fourel G, Couturier J, Wei Y, Apiou F, Tiollais P, Buendia MA. Evidence for long-range oncogene activation by hepadnavirus insertion [J]. EMBO J, 1994, 13 (11): 2526-2534.
[42] Hsu T, Möröy T, Etiemble J, Louise A, Trépo C, Tiollais P, Buendia MA. Activation of c-myc by woodchuck hepatitis virus insertion in hepatocellular carcinoma [J]. Cell, 1988, 55(4):627-635.
[43] Popper H, Roth L, Purcell RH, Tennant BC, Gerin JL. Hepatocarcinogenicity of the woodchuck hepatitis virus [J]. Proc Natl Acad Sci USA, 1987, 84 (3):866-870.
[44] Snyder RL, Tyler G, Summers J. Chronic hepatitis and hepatocellular carcinoma associated with woodchuck hepatitis virus [J]. Am J Pathol, 1982, 107 (3):422-425.
[45] Jacob JR, Sterczer A, Toshkov IA, Yeager AE, Korba BE, Cote PJ, Buendia MA, Gerin JL, Tennant BC. Integration of woodchuck hepatitis and N-myc rearrangement determine size and histologic grade of hepatic tumors [J]. Hepatology, 2004, 39(4):1008-1016.
[46] Buckwold VE, Beer BE, Donis RO. Bovine viral diarrhea virus as a surrogate model of hepatitis C virus for the evaluation of antiviral agents [J]. Antiviral Res, 2003, 60 (1): 1-15.
[47] Chambers TJ, Hahn CS, Galler R, Rice CM. Flavivirus genome organization, expression, and replication [J]. Annu Rev Microbiol, 1990, 44: 649-688.
[48] Neyts J, Leyssen P, De Clercq E. Infections with flaviviridae [J]. Verh K Acad Geneeskd Belg, 1999, 61(6): 661-699.
[49] Porter BF, Goens SD, Brasky KM, Hubbard GB. A case report of hepatocellular carcinoma and focal nodular hyperplasia with a myelolipoma in two chimpanzees and a review of spontaneous hepatobiliary tumors in non-human primates [J]. J Med Primatol, 2004, 33 (1): 38-47.
[50] Chisari FV, Klopchin K, Moriyama T, Pasquinelli C, Dunsford HA, Sell S, Pinkert CA, Brinster RL, Palmiter RD. Molecular pathogenesis of hepatocellular carcinoma in hepatitis B virus transgenic mice [J]. Cell, 1989, 59(6):1145-1156.
[51] Dunsford HA, Sell S, Chisari FV. Hepatocarcinogenesis due to chronic liver cell injury in hepatitis B virus transgenic mice [J]. Cancer Res, 1990, 50 (11):3400-3407.
[52] Yoakum GH, Korba BE, Lechner JF, Tokiwa T, Gazdar AF, Seeley T, Siegel M, Leeman L, Autrup H, Harris CC. High frequency transfection and cytopathology of the hepatitis B virus core antigen gene in human cells [J]. Science, 1983, 222(4622):385-389.
[53] Guidotti LG, Matzke B, Schaller H, Chisari FV. High level hepatitis B virus replication in transgenic mice [J]. J Virol, 1995, 69 (10): 6158-6169.
[54] Hoofnagle JH, Shafritz DA, Popper H. Chronic type B hepatitis and the “healthy” HBsAg carrier state [J]. Hepatology, 1987, 7 (4): 758-763.
[55] Sureau C, Romet-Lemonne JL, Mullins JI, Essex M. Production of hepatitis B virus by a differentiated human hepatoma cell line after transfection with cloned circular HBV DNA [J]. Cell, 1986, 7(1):37-47.
[56] Sells MA, Chen ML, Acs G. Production of hepatitis B virus particles in HepG2 cells transfected with cloned hepatitis B virus DNA [J]. Proc Natl Acad Sci USA, 1987, 84 (4): 1005-1009.
[57] Ando K, Moriyama T, Guidotti LG, Wirth S, Schreiber RD, Schlicht HJ, Huang SN, Chisari FV. Mechanisms of class 1 restricted immunopathology. A transgenic mouse model of fulminant hepatitis [J]. J Exp Med, 1993,178(5):1541-1544.
[58] Feitelson MA. Hepatitis B virus gene products as immunological targets in chronic infection [J]. Mol Biol Med, 1989, 6 (5): 367-393.
[59] Beasley RP, Hwang LY, Lin CC, Chien CS. Hepatocellular carcinoma and HBV: A prospective study of 22,707 men in Taiwan [J]. Lancet, 1981, 2 (8256):1129-1133.
[60] Feitelson MA. Hepatocellular injury in hepatitis B and C virus infections [M]. In: Feitelson MA, Zern MA. eds. Clinics in Laboratory Medicine: Hepatitis and Chronic Liver Disease. Philadelphia: WB Saunders Co., 1996, 307-324.
[61] Ferrari C, Penna A, Giuberti T, Tong MJ, Ribera E, Fiaccadori F, Chisari FV. Intrahepatic, nucleocapsid antigen-specific T cells in chronic active hepatitis B [J]. J Immunol, 1987, 139(6):2050-2058.
[62] Marinos G, Torre F, Chokshi S, Hussain M, Clarke BE, Rowlands DJ, Eddleston AL, Naoumov NV, Williams R. Induction of T-helper cell response to hepatitis B core antigen in chronic hepatitis B: A major factor in activation of the host immune response to the hepatitis B virus [J]. Hepatology, 1995, 22(4 Pt 1): 1040-1049.
[63] Bianchi L, Gudat F. Immunopathology of hepatitis B [M]. In: Popper H, Schaffner F. eds. Progress in Liver Disease. Vol. 6. San Francisco: Grune and Stratton, 1979, 371-392.
[64] Moriyama T, Guilhot S, Klopchin K, Moss B, Pinkert CA, Palmiter RD, Brinster RL, Kanagawa O, Chisari FV. Immunobiology and pathogenesis of hepatocellular injury in hepatitis B virus transgenic mice [J]. Science, 1990, 248(4953):361-364.
[65] Larkin J, Clayton M, Sun B, Perchonock CE, Morgan JL, Siracusa LD, Michaels FH, Feitelson MA. Hepatitis B virus transgenic mouse model of chronic liver disease [J]. Nat Med, 1999, 5(8):907-912.
[66] Chisari FV, Ferrari C. Hepatitis B virus immunopathogenesis [J]. Annu Rev Immunol, 1995, 13: 29-60.
[67] Guidotti LG, Ishikawa T, Hobbs MV, Matzke B, Schreiber R, Chisari FV. Intracellular inactivation of the hepatitis B virus by cytotoxic T lymphocytes [J]. Immunity, 1996, 4 (1):25-36.
[68] Guidotti LG, Ando K, Hobbs MV, Ishikawa T, Runkel L, Schreiber RD, Chisari FV. Cytotoxic T lymphocytes inhibit hepatitis B virus gene expression by a noncytolytic mechanism in transgenic mice [J]. Proc Natl Acad Sci USA, 1994, 91(9):3764-3768.
[69] Guidotti LG, Borrow P, Hobbs MV, Matzke B, Gresser I, Oldstone MB, Chisari FV. Viral cross talk: Intracellular inactivation of the hepatitis B virus during an unrelated viral infection of the liver [J]. Proc Natl Acad Sci USA, 1996, 93(10): 4589-4594.
[70] Guidotti LG, Guilhot S, Chisari FV. Interleukin-2 and interferon alpha/beta downregulate hepatitis B virus gene expression in vivo by tumor necrosis factor dependent and independent pathways [J]. J Virol, 1994, 68 (3):1265-1270.
[71] Heise T, Guidotti LG, Cavanaugh VJ, Chisari FV. Hepatitis B virus RNA binding proteins associated with cytokine induced clearance of viral RNA from the liver of transgenic mice [J]. J Virol, 1999, 73 (1): 474-481.
[72] Sitia G, Isogawa M, Iannacone M, Campbell IL, Chisari FV, Guidotti LG. MMPs are required for recruitment of antigen nonspecific mononuclear cells into the liver by CTLs [J]. J Clin Invest, 2004, 113 (8): 1158-1167.
[73] Baron JL, Gardiner L, Nishimura S, Shinkai K, Locksley R, Ganem D. Activation of a nonclassical NKT cell subset in a transgenic mouse model of hepatitis B virus infection [J]. Immunity, 2002, 16 (4): 583-594.
[74] Collier J, Heathcote J. Hepatitis C viral infection in the immunosuppressed patient [J]. Hepatology, 1998, 27 (1): 2-6.
[75] Thomas DL, Shih JW, Alter HJ, Vlahov D, Cohn S, Hoover DR, Cheung L, Nelson KE. Effect of human immuno-deficiency virus on hepatitis C virus infection among injection drug users [J]. J Infect Dis, 1996, 174(4):690-695.
[76] Zhong J, Gastaminza P, Chung J, Stamataki Z, Isogawa M, Cheng G, McKeating JA, Chisari FV. Persistent hepatitis C virus infection in vitro: coevolution of virus and host [J]. J Virol, 2006, 80(22):11082-11093.
[77] Walters KA, Syder AJ, Lederer SL, Diamond DL, Paeper B, Rice CM, Katze MG. Genomic analysis reveals a potential role for cell cycle perturbation in HCV-mediated apoptosis of cultured hepatocytes [J]. PLoS Pathog, 2009, 5 (1): e1000269.
[78] Bronkhorst CM, ten Kate FJ. Liver histology in hepatitis C [J]. Curr Stud Hematol Blood Transfus, 1998, 62: 119-134.
[79] Diepolder HM, Hoffmann RM, Gerlach JT, Zachoval R, Jung MC, Pape GR. Immunopathogenesis of HCV infection [J]. Curr Stud Hematol Blood Transfus, 1998, 62: 135-151.
[80] Bradley DW. Studies of non-A, non-B hepatitis and characterization of the hepatitis C virus in chimpanzees [J]. Curr Top Microbiol Immunol, 2000, 242: 1-23.
[81] Herzer K, Sprinzl MF, Galle PR. Hepatitis viruses: live and let die [J]. Liver Int, 2007, 27 (3): 293-301.
[82] Kremsdorf D, Brezillon N. New animal models for hepatitis C viral infection and pathogenesis studies [J]. World J Gastroenterol, 2007, 13 (17):2427-2435.
[83] Palmiter RD, Brinster RL. Transgenic mice[J]. Cell, 1985, 41: 343-345.
[84] Babinet C, Farza H, Morello D, Hadchouel M, Pourcel C. Specific expression of hepatitis B surface antigen (HBsAg) in transgenic mice [J]. Science, 1985, 230 (4730): 1160-1163.
[85] Chisari FV, Pinkert CA, Milich DR, Filippi P, McLachlan A, Palmiter RD, Brinster RL. A transgenic mouse model of the chronic hepatitis B surface antigen carrier state [J]. Science, 1985, 230 (4730): 1157-1160.
[86] Burk RD, DeLoia JA, elAwady MK, Gearhart JD. Tissue preferential expression of the hepatitis B virus (HBV) surface antigen gene in two lines of HBV transgenic mice [J]. J Virol, 1988, 62 (2): 649-654.
[87] Araki K, Miyazaki J, Hino O, Tomita N, Chisaka O, Matsubara K, Yamamura K. Expression and replication of hepatitis B virus genome in transgenic mice [J]. Proc Natl Acad Sci USA, 1989, 86 (1):207-211.
[88] Farza H, Hadchouel M, Scotto J, Tioillais P, Babinet C, Pourcel C. Replication and gene expression of hepatitis B virus in a transgenic mouse that contains the complete viral genome [J]. J Virol, 1988, 62 (11):4144-4152.
[89] Chisari FV, Filippi P, McLachlan A, Milich DR, Riggs M, Lee S, Palmiter RD, Pinkert CA, Brinster RL. Expression of hepattiis B virus large envelope polypeptide inhibits hepatitis B surface antigen secretion in transgenic mice [J]. J Virol, 1986, 60(3):880-887.
[90] Kim CM, Koike K, Saito I, Miyamura T, Jay G. HBx gene of hepatitis B virus induces liver cancer in transgenic mice [J]. Nature, 1991, 351 (6324):317-320.
[91] Koike K, Moriya K, Iino S, Yotsuyanagi H, Endo Y, Miyamura T, Kurokawa K. High-level expression of hepatitis B virus HBx gene and hepatocarcinogenesis in transgenic mice [J]. Hepatology, 1994, 19 (4):810-819.
[92] Ueda H, Ullrich SJ, Gangemi JD, Kappel CA, Ngo L, Feitelson MA, Jay G. Functional inactivation but not structural mutation of p53 causes liver cancer [J]. Nat Genet, 1995, 9(1): 41-47.
[93] Lee TH, Finegold MJ, Shen RF, DeMayo JL, Woo SL, Butel JS. Hepatitis B virus transactivator X protein is not tumorigenic in transgenic mice [J]. J Virol, 1990, 64 (12): 5939-5947.
[94] Reifenberg K, Löhler J, Pudollek HP, Schmitteckert E, Spindler G, Köck J, Schlicht HJ. Long-term expression of the hepatitis B virus core-e- and X-proteins does not cause pathologic changes in transgenic mice [J]. J Hepatol, 1997, 26(1): 119-130.
[95] Yoo YD, Ueda H, Park K, Flanders KC, Lee YI, Jay G, Kim SJ. Regulation of transforming growth factor-beta 1 expression by the hepatitis B virus (HBV) X transactivator. Role in HBV pathogenesis [J]. J Clin Invest, 1996, 97 (2):388-395.
[96] Akhurst RJ. TGF-beta antagonists: why suppress a tumor suppressor [J]? J Clin Invest, 2002, 109 (12):1533-1536.
[97] Murata M, Matsuzaki K, Yoshida K, Sekimoto G, Tahashi Y, Mori S, Uemura Y, Sakaida N, Fujisawa J, Seki T, Kobayashi K, Yokote K, Koike K, Okazaki K. Hepatitis B virus X protein shifts human hepatic transforming growth factor (TGF)-beta signaling from tumor suppression to oncogenesis in early chronic hepatitis B [J]. Hepatology, 2009, 49(4):1203-1217.
[98] Yu DY, Moon HB, Son JK, Jeong S, Yu SL, Yoon H, Han YM, Lee CS, Park JS, Lee CH, Hyun BH, Murakami S, Lee KK. Incidence of hepatocellular carcinoma in transgenic mice expressing the hepatitis B virus X-protein [J]. J Hepatol, 1999, 31(1):123-132.
[99] Koo JS, Seong JK, Park C, Yu DY, Oh BK, Oh SH, Park YN. Large liver cell dysplasia in hepatitis B virus x transgenic mouse liver and human chronic hepatitis B virus-infected liver [J]. Intervirology, 2005, 48 (1):16-22.
[100] Wang W, London WT, Feitelson MA. Hepatitis B x antigen in hepatitis B virus carrier patients with liver cancer [J]. Cancer Res, 1991, 51 (18): 4971-4977.
[101] Wang W, London WT, Lega L, Feitelson MA. HBxAg in liver from carrier patients with chronic hepatitis and cirrhosis [J]. Hepatology, 1991, 14 (1): 29-37.
[102] Lee YI, Hwang JM, Im JH, Lee YI, Kim NS, Kim DG, Yu DY, Moon HB, Park SK. Human hepatitis B virus-X protein alters mitochondrial function and physiology in human liver cells [J]. J Biol Chem, 2004, 279(15):15460-15471.
[103] Cote PJ, Korba BE, Steinberg H, Ramirez-Mejia C, Baldwin B, Hornbuckle WE, Tennant BC, Gerin JL. Cyclosporin A modulates the course of woodchuck hepatitis virus infection and induces chronicity [J]. J Immunol, 1991, 146(9):3138-3144.
[104] Takahashi H, Fujimoto J, Hanada S, Isselbacher KJ. Acute hepatitis in rats expressing human hepatitis B virus transgenes [J]. Proc Natl Acad Sci USA, 1995, 92 (5):1470-1474.
[105] Nakamoto Y, Guidotti LG, Kuhlen CV, Fowler P, Chisari FV. Immune pathogenesis of hepatocellular carcinoma [J]. J Exp Med, 1998, 188 (2): 341-350.
[106] Ray MB. Hepatitis B Virus Antigens in Tissues [M]. Baltimore, MD: University Park Press, 1979.
[107] Kajino K, Jilbert AR, Saputelli J, Aldrich C, Cullen J, Mason WS. Woodchuck hepatitis virus infections: very rapid recovery after a prolonged viremia and infection of virtually every hepatocyte [J]. J Virol, 1994, 68 (9):5792-5803.
[108] Gilles PN, Fey G, Chisari FV. Tumor necrosis factor alpha negatively regulates hepatitis B virus gene expression in transgenic mice [J]. J Virol, 1992, 66 (6): 3955-3960.
[109] Guilhot S, Guidotti LG, Chisari FV. Interleukin-2 downregulates hepatitis B virus gene expression in transgenic mice by a post-transcriptional mechanism [J]. J Virol, 1993, 67 (12):7444-7449.
[110] Tsai LV, Guidotti LG, Ishkawa T, Chisari FV. Posttranscriptional clearance of hepatitis B virus RNA by cytotoxic T lymphocytes-activated hepatocytes [J]. Proc Natl Acad Sci USA, 1995, 92: (26):12398-12402.
[111] Cavanaugh VJ, Guidotti LG, Chisari FV. Interleukin-12 inhibits hepatitis B virus replication in transgenic mice [J]. J Virol, 1997, 71 (4): 3236-3243.
[112] Liaw YF. Role of hepatitis C virus in dual and triple hepatitis virus infection [J]. Hepatology, 1995, 22 (4Pt1): 1101-1108.
[113] Xuan B, Qian Z, Hong J, Huang W. EsiRNAs inhibit hepatitis B virus replication in mice model more efficiently than synthesized siRNAs [J]. Virus Res, 2006, 118 (1-2): 150-155.
[114] Chen CC, Ko TM, Ma HI, Wu HL, Xiao X, Li J, Chang CM, Wu PY, Chen CH, Han JM, Yu CP, Jeng KS, Hu CP, Tao MH. Long-term inhibition of hepatitis B virus in transgenic mice by double-stranded adeno-associated virus 8-delivered short hairpin RNA [J]. Gene Ther, 2007, 14(1): 11-19.
[115] Grimm D, Wang L, Lee JS, Schürmann N, Gu S, Börner K, Storm TA, Kay MA. Argonaute proteins are key determinants of RNAi efficacy, toxicity, and persistence in the adult mouse liver [J]. J Clin Invest, 2010, 120(9):3106-3119.
[116] Kamkolkar M, Clayton MM, Zhang SM, Black P, Schinazi RF, Feitelson MA. Evaluation of therapies for hepatitis B virus in the HBV transgenic SCID mouse model [M]. In: Schinazi RF, Rice C, Sommadossi P. eds. Frontiers in Viral Hepatitis. Amsterdam: Elsevier B.V., 2003: 211-222.
[117] Chen H, Pai SB, Hurwitz SJ, Chu CK, Glazkova Y, McClure HM, Feitelson M, Schinazi RF. Antiviral activity and pharmacokinetics of the antiviral agent 1-(2,3,-dideoxy-2-fluoro-beta-L-glyceropent-2-enofuranosyl) cytosine [J]. Antimicrob Agents Chemother, 2003, 47(6):1922-1928.
[118] Furukawa S, Akbar SM, Hasebe A, Horiike N, Onji M. Production of hepatitis B surface antigen-pulsed dendritic cells from immunosuppressed murine hepatitis B virus carrier: evaluation of immunogenicity of antigen-pulsed dendritic cells in vivo [J]. Immunobiology, 2004, 209 (7): 551-557.
[119] Koike K, Moriya K, Ishibashi K, Matsuura Y, Suzuki T, Saito I, Iino S, Kurokawa K, Miyamura T. Expression of hepatitis C virus envelope proteins in transgenic mice [J]. J Gen Virol, 1995, 76 ( Pt 12):3031-3038.
[120] Koike K, Moriya K, Ishibashi K, Yotsuyanagi H, Shintani Y, Fujie H, Kurokawa K, Matsuura Y, Miyamura T. Sialadenitis histologically resembling Sjogren syndrome in mice transgenic for hepatitis C virus envelope genes [J]. Proc Natl Acad Sci USA, 1997, 94 (1):233-236.
[121] Haddad J, Deny P, Munz-Gotheil C, Ambrosini JC, Trinchet JC, Pateron D, Mal F, Callard P, Beaugrand M. Lymphocytic sialadenitis of Sjogren’s syndrome associated with chronic hepatitis C virus liver disease [J]. Lancet, 1992, 339(8789):321-323.
[122] Pasquinelli C, Shoenberger JM, Chung J, Chang KM, Guidotti LG, Selby M, Berger K, Lesniewski R, Houghton M, Chisari FV. Hepatitis C virus core and E2 protein expression in transgenic mice [J]. Hepatology, 1997, 25 (3):719-727.
[123] Kawamura T, Furusaka A, Koziel MJ, Chung RT, Wang TC, Schmidt EV, Liang TJ. Transgenic expression of hepatitis C virus structural proteins in the mouse [J]. Hepatology, 1997, 25(4):1014-1021.
[124] Honda A, Arai Y, Hirota N, Sato T, Ikegaki J, Koizumi T, Hatano M, Kohara M, Moriyama T, Imawari M, Shimotohno K, Tokuhisa T. Hepatitis C virus structural proteins induce liver cell injury in transgenic mice [J]. J Med Virol, 1999, 59(3):281-289.
[125] Encke J, Geissler M, Stremmel W, Wands JR. DNA-based immunization breaks tolerance in a hepatitis C virus transgenic mouse model [J]. Hum Vaccin, 2006, 2 (2): 78-83.
[126] Wakita T, Taya C, Katsume A, Kato J, Yonekawa H, Kanegae Y, Saito I, Hayashi Y, Koike M, Kohara M. Efficient conditional transgene expression in hepatitis C virus cDNA transgenic mice mediated by the Cre/loxP system [J]. J Biol Chem, 1998, 273(15):9001-9006.
[127] Moriya K, Yotsuyanagi H, Shintani Y, Fujie H, Ishibashi K, Matsuura Y, Miyamura T, Koike K. Hepatitis C virus core protein induced hepatic steatosis in transgenic mice [J]. J Gen Virol, 1997, 78 (Pt7):1527-1531.
[128] Moriya K, Fujie H, Shintani Y, Yotsuyanagi H, Tsutsumi T, Ishibashi K, Matsuura Y, Kimura S, Miyamura T, Koike K. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice [J]. Nat Med, 1998, 4(9):1065-1067.
[129] Chang J, Yang SH, Cho YG, Hwang SB, Hahn YS, Sung YC. Hepatitis C virus from two different genotypes has an oncogenic potential but is not sufficient for transforming primary rat embryo fibroblasts in cooperation with the H-ras oncogene [J]. J Virol, 1998, 72 (4): 3060-3065.
[130] Ray RB, Lagging LM, Meyer K, Steele R, Ray R. Transcriptional regulation of cellular and viral promoters by the hepatitis C virus core protein [J]. Virus Res, 1995, 37 (3): 209-220.
[131] Lemon SM, Lerat H, Weinman SA, Honda M. A transgenic mouse model of steatosis and hepatocellular carcinoma associated with chronic hepatitis C virus infection in humans [J]. Trans Am Clin Climatol Assoc, 2000, 111: 146-156; discussion 156-157.
[132] Matsuda J, Suzuki M, Nozaki C, Shinya N, Tashiro K, Mizuno K, Uchinuno Y, Yamamura K. Transgenic mouse expressing a full length hepatitis C virus cDNA [J]. Jpn J Cancer Res, 1998, 89(2):150-158.
[133] Keasler VV, Lerat H, Madden CR, Finegold MJ, McGarvey MJ, Mohammed EM, Forbes SJ, Lemon SM, Hadsell DL, Grona SJ, Hollinger FB, Slagle BL. Increased liver pathology in hepatitis C virus transgenic mice expressing the hepatitis B virus X protein [J]. Virology, 2006, 347(2):466-475.
[134] Ilan E, Burakova T, Dagan S, Nussbaum O, Lubin I, Eren R, Ben-Moshe O, Arazi J, Berr S, Neville L, Yuen L, Mansour TS, Gillard J, Eid A, Jurim O, Shouval D, Reisner Y, Galun E. The hepatitis B virus trimera mouse: a model for human HBV infection and evaluation of anti-HBV therapeutic agents [J]. Hepatology, 1999, 29(2):553-562.
[135] Ohashi K, Marion PL, Nakai H, Meuse L, Cullen JM, Bordier BB, Schwall R, Greenberg HB, Glenn JS, Kay MA. Sustained survival of human hepatocytes in mice: a model for in vivo infection with human hepatitis B and hepatitis delta viruses [J]. Nat Med, 2000, 6(3):327-331.
[136] Brown JJ, Parashar B, Moshage H, Tanaka KE, Engelhardt D, Rabbani E, Roy-Chowdhury N, Roy-Chowdhury J. A long-term hepatitis B viremia model generated by transplanting nontumorigenic immortalized human hepatocytes in Rag-2-deficient mice [J]. Hepatology, 2000, 31(1):173-181.
[137] Sandgren EP, Palmiter RD, Heckel JL, Daugherty CC, Brinster RL, Degen JL. Complete hepatic regeneration after somatic deletion of an albumin-plasminogen activator transgene [J]. Cell, 1991, 66 (2): 245-256.
[138] Petersen J, Dandri M, Gupta S, Rogler CE. Liver repopulation with xenogenic hepatocytes in B and T cell deficient mice leads to chronic hepadnavirus infection and clonal growth of hepatocellular carcinoma [J]. Proc Natl Acad Sci USA, 1998, 95 (1): 310-315.
[139] Dandri M, Burda MR, Török E, Pollok JM, Iwanska A, Sommer G, Rogiers X, Rogler CE, Gupta S, Will H, Greten H, Petersen J. Repopulation of mouse liver with human hepatocytes and in vivo infection with hepatitis B virus [J]. Hepatology, 2001, 33(4): 981-988.
[140] Mercer DF, Schiller DE, Elliott JF, Douglas DN, Hao C, Rinfret A, Addison WR, Fischer KP, Churchill TA, Lakey JR, Tyrrell DL, Kneteman NM. Hepatitis C virus replication in mice with chimeric human livers [J]. Nat Med, 2001, 7(8):927-933.
[141] Meuleman P, Libbrecht L, De Vos R, de Hemptinne B, Gevaert K, Vandekerckhove J, Roskams T, Leroux-Roels G. Morphological and biochemical characterization of a human liver in a uPA-SCID mouse chimera [J]. Hepatology, 2005, 41(4):847-856.
[142] Tateno C, Yoshizane Y, Saito N, Kataoka M, Utoh R, Yamasaki C, Tachibana A, Soeno Y, Asahina K, Hino H, Asahara T, Yokoi T, Furukawa T, Yoshizato K. Near completely humanized liver in mice shows human-type metabolic responses to drugs [J]. Am J Pathol, 2004,165(3):901-912.
[143] Kneteman NM, Howe AY, Gao T, Lewis J, Pevear D, Lund G, Douglas D, Mercer DF, Tyrrell DL, Immermann F, Chaudhary I, Speth J, Villano SA, O'Connell J, Collett M. HCV796: A selective nonstructural protein 5B polymerase inhibitor with potent anti-hepatitis C virus activity in vitro, in mice with chimeric human livers, and in humans infected with hepatitis C virus [J]. Hepatology, 2009, 49(3):745-752.
[144] Kamiya N, Iwao E, Hiraga N, Tsuge M, Imamura M, Takahashi S, Miyoshi S, Tateno C, Yoshizato K, Chayama K. Practical evaluation of a mouse with chimeric human liver model for hepatitis C virus infection using an NS3-4A protease inhibitor [J]. J Gen Virol, 2010, 91(Pt 7):1668-1677.
[145] Meuleman P, Leroux-Roels G. The human liver-uPA-SCID mouse: A model for the evaluation of antiviral compounds against HBV and HCV [J]. Antiviral Res, 2008, 80 (3): 231-238.
[146] Vanwolleghem T, Libbrecht L, Hansen BE, Desombere I, Roskams T, Meuleman P, Leroux-Roels G. Factors determining successful engraftment of hepatocytes and susceptibility to hepatitis B and C virus infection in uPA-SCID mice [J]. J Hepatol, 2010, 53(3):468-476.
[147] Ladner SK, Otto MJ, Barker CS, Zaifert K, Wang GH, Guo JT, Seeger C, King RW. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: a novel system for screening potential inhibitors of HBV replication [J]. Antimicrob Agents Chemother, 1997, 41(8):1715-1720.
[148] Feitelson MA, Clayton MM, Sun B, Schinazi RF. Development of a novel mouse model to evaluate drug candidates against hepatitis B virus [J]. Antivir Chem Chemother, 2007, 18 (4): 213-223.
[149] Guévin C, Lamarre A, Labonté P. Novel HCV replication mouse model using human hepatocellular carcinoma xenografts [J]. Antiviral Res, 2009, 84 (1): 14-22.
[150] Zhu Q, Weiner AJ. A hepatitis C virus xenograft mouse efficacy model [J]. Methods Mol Biol, 2009, 511: 323-331.
[151] Sun BS, Pan J, Clayton MM, Liu J, Yan X, Matskevich AA, Strayer DS, Gerber M, Feitelson MA. Hepatitis C virus replication in stably transfected HepG2 cells promotes hepatocellular growth and tumorigenesis [J]. J Cell Physiol, 2004, 201(3): 447-458.
[152] Wu GY, Konishi M, Walton CM, Olive D, Hayashi K, Wu CH. A novel immunocompetent rat model of HCV infection and hepatitis [J]. Gastroenterology, 2005, 128 (5):1416-1423.
[153] Hoofnagle JH. Hepatitis C: the clinical spectrum of disease [J]. Hepatology, 1997, 26 (3 Suppl 1):15S-20S.
