
高效液相色谱法同步测定食品中5种色素含量
2012-01-19 03:28徐丽红王建清
浙江农业科学 2012年3期
张 玉,王 伟,徐丽红,王建清
(浙江省农业科学院 农产品质量标准研究所,浙江 杭州 310021)
碱性嫩黄O、酸性橙II、碱性橙II、罗丹明B、对位红均为偶氮类工业染料。广泛用于纺织、皮革、造纸、橡胶、塑料、涂料、化妆品、木材加工等,它们具有一定的毒性,部分还有致癌性,因此禁止作为食品添加剂使用。但由于工业染料色泽鲜艳、着色稳定且价格低廉,一些不法商贩将其用于食品生产与加工,严重危害了消费者的身体健康[1-2]。
目前对于食品中违禁色素的检测方法已有一定研究,主要有薄层色谱法、液相色谱法和高效液相色谱-串联质谱法[3-9]。但大多数研究集中在对1~2种物质的检测。为此,我们通过固相萃取技术结合高效液相色谱法,建立食品中碱性嫩黄O、酸性橙II、碱性橙 II、罗丹明 B、对位红同步测定方法。现将有关结果报道如下。
1 材料与方法
1.1 检测样品、试剂与仪器
检测样品有辣椒酱和腐竹。
试剂有标准品碱性嫩黄O(纯度95%)、酸性橙II(纯度98%)、碱性橙 II(纯度98%)、罗丹明B(纯度92%)、对位红 (纯度96%);甲醇和乙腈 (色谱纯);乙醇 (分析纯)。
仪器有戴安Ultimate-3000高效液相色谱仪,带紫外检测器;超声波清洗器;旋转蒸发仪;离心机;HLB固相萃取小柱 (200 mg,3 mL)。
1.2 标准溶液配制
准确称取碱性嫩黄 O、酸性橙 II、碱性橙 II、罗丹明B、对位红标准品各0.050 0 g,分别置于50 mL容量瓶中,甲醇溶解并定容至刻度,得到浓度为1.0 g·L-1的标准贮备液。
分别吸取5.0 mL 5种标准储备液于100 mL容量瓶中,混匀后,用甲醇定容至刻度,得到浓度为50 mg·L-1的混合标准溶液。再移取50 mg·L-1的混合标准溶液0.1,0.2,1.0,2.0,分别置于10 mL容量瓶中,用流动相定容,此标准系列质量浓度为0.5,1.0,5.0,10.0 mg·L-1。
1.3 样品前处理
称取5.0 g捣碎样品置于50 mL离心管中,加入30 mL乙醇和乙腈混合液 (V/V为80∶20),高速均质1 min,超声萃取20 min,离心,移出上清液,残渣用30 mL乙醇和乙腈混合液再提取1次,合并上清液,用旋转蒸发仪浓缩近干后,加入2 mL乙醇和乙腈混合液溶解,再加入8 mL水混匀为待净化液。
依次用5 mL甲醇和5 mL水活化固相萃取柱,将待净化液全部上柱。依次用10 mL甲醇和10 mL正己烷洗脱,接收并合并洗脱液,用旋转蒸发仪浓缩近干后,加2 mL乙醇和乙腈混合液溶解,然后定容。
1.4 色谱分析条件
流动相:甲醇、乙酸铵缓冲液和乙腈梯度洗脱(表1),色谱柱:C18,4.6 mm×250 mm(5μm);流速:1.0 mL·min-1;柱温:25℃;进样体积:20μL;波长450 nm,480 nm,530 nm。
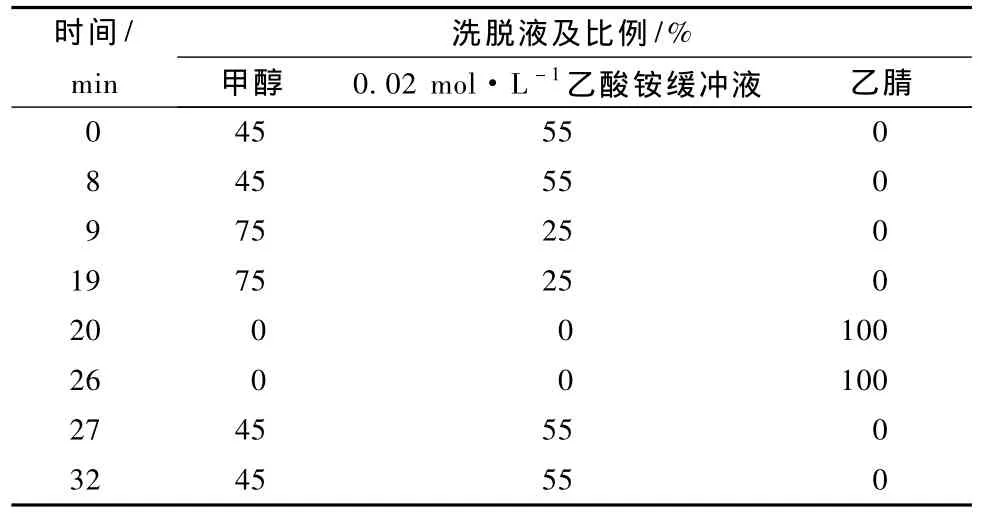
表1 色谱柱洗脱的流动相梯度
2 结果与分析
2.1 检测波长的确定
对5种色素的标准品进行光谱扫描,发现5种色素的最大吸收波长分别为:碱性嫩黄 O,435 nm;酸性橙II,480 nm;碱性橙II,430 nm;罗丹明B,530 nm;对位红,480 nm。
综合考虑几种色素的最大吸收波长,进行分段波长检测,即0~15 min检测波长为450 nm,15~20 min检测波长为530 nm,20~27 min检测波长为480 nm。
2.2 液相色谱条件的确定
根据样品分离情况,优化流动相流速及配比,确定用甲醇、乙酸铵缓冲液和乙腈梯度洗脱。从图1-2看出,样品中碱性嫩黄O、酸性橙II、碱性橙II、罗丹明 B、对位红能够完全分离,得到理想色谱。

图1 5种工业染料标准物质色谱
2.3 提取液的选择

图2 腐竹样品添标色谱
采用不同的提取液对样品中的色素进行提取,结果 (表2)4种提取液对样品中色素的提取回收率80.2%~101.2%,其中乙醇+乙腈混合液提取后,不同色素回收率为85.1%~98.8%,差异较小,因此确定此混合溶剂作为提取液。
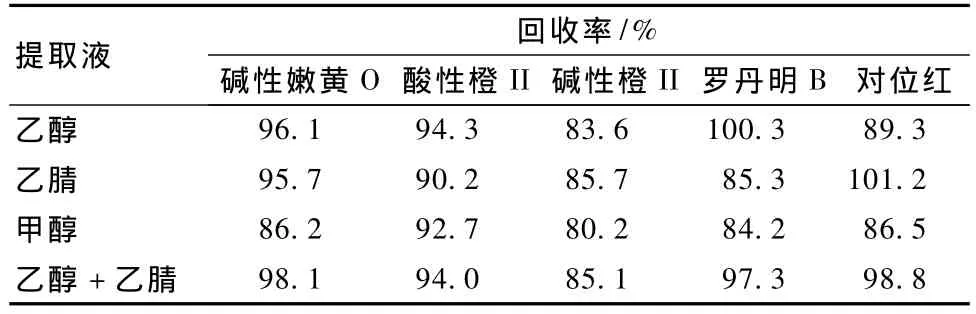
表2 不同提取液提取5种色素的回收率
2.4 固相萃取条件筛选
由于样品提取液中含有一定的杂质,会干扰色素的色谱分离,因此采用固相萃取柱对样品进行净化。先采用标准物质进行固相萃取条件筛选。表3表明,单用洗脱液1(10 mL甲醇)洗脱,对位红的回收率较差,而用洗脱液1洗脱后再用洗脱液2(10 mL正己烷)洗脱,5种色素均能得到较好回收率。

表3 不同洗脱液对5种色素固相萃取回收率的影响
2.5 方法的线性及检出限
将配制好的系列标准工作液上机测定,以浓度为横坐标,以峰面积为纵坐标,绘制标准曲线。在浓度为0.5~50 mg·L-1的浓度范围里,5种标准物质具有良好的线性相关 (表4)。方法的检出限为 0.03 ~0.2 mg·kg-1。
2.6 方法回收率
采用辣椒酱和腐竹进行不同添加浓度的样品回收率实验,结果表明,该方法具有良好的准确度和重复性,5种违禁色素回收率为82.5% ~96.9%,相对标准偏差 (RSD)为1.0% ~3.5%(表5)。

表4 方法的线性及检出限

表5 不同食品中5种色素的回收率
3 小结
本研究建立了高效液相色谱同时测定食品中5种色素含量的方法。该方法线性关系良好,在0.5~50 mg·L-1范围内,相关系数为 0.999 8~0.999 9,方法的回收率为82.5% ~96.9%,精密度RSD为1.0%~3.5%,具有较高的准确度和良好的重现性,可作为同时进行食品中碱性嫩黄O、酸性橙II、碱性橙II、罗丹明B、对位红含量测定的方法。
[1] 肖义夫,廖百森.食品中非食用色素及其检测方法研究进展 [J].现代预防医学,2008,35(16):3159-3164.
[2] 卢士英,邹明强.食品中常见的非食用色素的危害与检测[J].食品及农药残留检测,2009(8):45-50.
[3] 张耀武,顾慧莹,路军辉.高效液相色谱法测定食品中的酸性橙Ⅱ [J].化学分析计量,2005,14(1):48-49.[4] 彭景龙.高效液相色谱法测定调味品中“对位红”含量 [J].中国调味品,2006(2):46-48.
[5] 陈春晓,刘红河,仲岳桐,等.食品中酸性橙Ⅱ的HPLC—MS/MS测定方法研究 [J].中国卫生检验杂志,2008,18(11):2209-2211.
[6] 铁晓威,黄百芬,任一平.RP-HPLC法测定染色黄鱼中的碱性橙含量 [J].中国卫生检验杂志,2004(1):63-64.
[7] 高洁,尹峰,何国亮,等.高效液相色谱法测定豆制品中的碱性嫩黄O[J].分析试验室,2008(增刊1):230-232.
[8] 林钦.高效液相色谱法同时测定豆制品中的碱性橙和碱性嫩黄O染料 [J].色谱,2007,25(5):776-777.
[9] 夏立娅,吴广臣,韩媛媛,等.食品中碱性橙、皂黄、丽春红2R等色素的同时薄层色谱定性分析 [J].食品工业科技,2009,30(6):296-297.
猜你喜欢
天津音乐学院学报(2022年2期)2022-07-28
家庭科学·新健康(2022年7期)2022-07-13
安徽化工(2022年2期)2022-04-12
表面技术(2022年1期)2022-02-12
蚌埠医学院学报(2020年11期)2020-12-17
——论传统对位教学两种体系的冲突
艺术探索(2020年1期)2020-05-08
音乐研究(2019年5期)2019-11-22
安徽科技学院学报(2018年3期)2018-09-18
伴侣(2018年8期)2018-08-23
分析化学(2017年12期)2017-12-25
