
连枷臂综合征的临床特征分析
2012-01-04 01:53王特孙新刚杨欢刘运海
中国全科医学 2012年32期
王特,孙新刚,杨欢,刘运海
连枷臂综合征的临床特征分析
王特,孙新刚,杨欢,刘运海
目的探讨连枷臂综合征(FAS)的临床特征。方法回顾性分析2010-06-01—2011-12-01我院神经内科收治的8例FAS患者的临床资料并总结其临床特征。结果8例患者主要临床特征以上肢近端的肌肉萎缩为主,中老年发病〔平均发病年龄(48.2±4.8)岁〕,男性多发(男女比例为7∶1)。首发症状以双侧上肢无力居多(62.5%),可伴有肌束颤动等。肌电图检查表现为广泛的(3个或3个节段以上)神经源性损害。肌肉活检标本8例均可见肌纤维萎缩,7例出现同型肌纤维群组化,3例可见靶纤维。结论FAS患者病情发展相对缓慢、生存时间更长,可能是肌萎缩侧索硬化的临床变异型,其发病可能与基因异常及种族相关。
连枷臂综合征;肌萎缩侧索硬化;运动神经元病
在研究肌萎缩侧索硬化(amyotrophic lateral sclerosis,ALS)的临床工作中,发现有一组病例,以对称性双上肢近端为主的显著萎缩和无力为突出表现,而双下肢、延髓受累轻微。此病首先于1886年由Vulpain报道,1888年Gowers以图片形式描述,1998年Hu将其命名为连枷臂综合征(flail arm syndrome,FAS)。FAS一般男性高发,患者生存期较长。本文通过对我院收治的8例FAS患者临床资料进行分析及总结相关文献,以进一步探讨FAS的临床特点。
1 资料与方法
1.1 一般资料收集2010-06-01—2011-12-01我院神经内科收治的8例FAS患者,诊断参照1998年修订的El-Escorial诊断标准[1],入选病例均有完整的临床诊断资料和随访记录。并根据临床表现、实验室和电生理检查结果排除其他疾病。
1.2 方法回顾性分析患者的一般资料、临床表现及实验室检查结果。
2 结果
2.1 患者一般资料8例FAS患者中男7例,女1例;年龄为41~68岁,平均(53.7±3.4)岁;发病年龄为38~67岁,平均(48.2±4.8)岁。
2.2 临床症状和体征8例患者均为慢性隐袭起病,就诊时病程最短为3个月,最长为4年,平均病程为2.1年。首发症状以双侧肌体无力居多,其中以双侧上肢无力为首发症状的有5例(62.5%),单侧上肢无力为首发症状的有2例(25.0%),咀嚼无力起病的有1例(12.5%)。患者均表现为不同程度的肌肉萎缩,以上肢近端的肌肉萎缩为主,由于该类患者三角肌、冈上下肌和小圆肌等明显萎缩,导致双侧上肢呈现特征性姿势,即肩部下沉、双上肢松弛垂放于躯干两侧(见图1、图2),有3例患者还出现舌肌萎缩和颤动。在其他伴随症状或体征中,出现肌束震颤的患者有6例,出现吞咽障碍的有1例。双上肢腱反射消失有1例,腱反射减低有7例;下肢腱反射亢进有3例,腱反射减低有2例,1例未引出,2例正常;下肢病理征阳性6例,阴性2例。

图1 患者肩部下沉、双上肢松弛垂放于躯干的两侧Figure 1 Patient keeps shoulders down and both upper limbs hang down on both sides of the body loosely

图2 患者三角肌、冈上下肌等明显萎缩Figure 2 Patient with obvious atrophy of deltoid muscle,supraspinatus and infraspinatus
2.3 实验室检查MRI检查:颅脑MRI正常3例,脑白质深部多发缺血灶3例,脑萎缩2例;5例患者出现轻度颈椎骨质、椎间盘或韧带异常改变,其中1例硬膜囊轻度受压,脊髓未见明显受压,其余均未出现脊髓及神经根异常改变。腰椎穿刺脑脊液压力、常规、生化检查正常。肺功能检查正常。肌电图检查:8例患者有广泛的(3个或3个节段以上)神经源性损害,主要表现为各肌肉组织出现失神经电位,轻收缩运动单位动作电位平均时限明显增宽、波幅增高,重收缩时受检肌呈单纯相或混合相;6例患者在静息状态下可见肌纤维颤动电位和肌束颤动电位;4例患者可见宽大运动单位电位;8例患者神经传导速度检查均未见异常;F波潜伏期正常。肌肉活检:光镜下8例肌肉活检标本均可见肌纤维萎缩,大部分萎缩肌纤维横断面带尖角或为扁平;均出现簇样分布小角状萎缩肌纤维(见图3),部分肌纤维肥大,其中1例患者病理切片局部可见肌纤维大群萎缩;肌纤维萎缩的程度群与群之间不相等,甚至每一小肌纤维群中每个肌纤维的萎缩程度也不尽一致,但病程越长,重度萎缩的肌纤维越多;7例出现同型肌纤维群组化,其中4例主要为Ⅱ型群组化(见图4),2例主要为Ⅰ型群组化,Ⅰ、Ⅱ型肌纤维同时出现群组化1例。NADH-TR染色3例可见靶纤维,部分萎缩的肌纤维深染(见图5)。
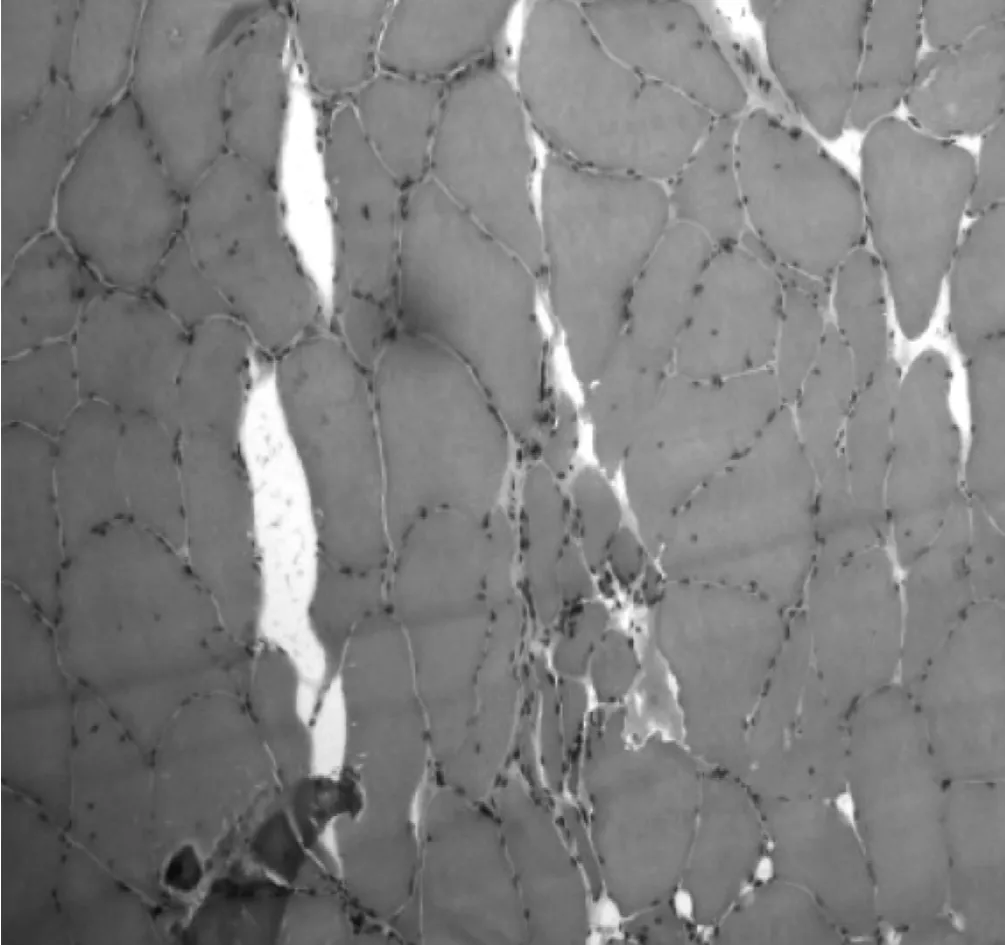
图3 光镜下小群肌萎缩(HE染色,10×20倍)Figure 3 Small group amyotrophia under light microscope(HE stain,10×20 times)

图4 光镜下见Ⅱ型肌纤维群组化(ATP染色,10×20倍,pH 4.3)Figure 4 TypeⅡmuscle fiber grouping seen under light microscope(ATP stain,10×20 times,pH 4.3)

图5 光镜下可见部分肌纤维酶活性减低(NADH-TR染色,10×20倍)Figure 5 Decrease of activity of some muscle fiber enzymes seen under light microscope(NADH-TR stain,10×20 times)
3 讨论
运动神经元病(motor neuron disease,MND)是一种常见的神经系统疾病,选择性侵犯脑和脊髓上、下运动神经元,起病隐袭,呈进行性发展,多数患者于起病后3~5年死于呼吸麻痹及其他并发症,ALS是其临床亚型。1998年Hu等[2]发现在MND患者中有10%的患者表现为严重的对称性以双上肢近端为主的肌无力和肌萎缩,但其他区域无或仅轻度受累,生存期相对较长。由于该类患者三角肌、冈上下肌、胸锁乳突肌和小圆肌等明显萎缩,导致双侧上肢呈现特征性姿势,即肩部下沉、双上肢松弛垂放于躯干的两侧,而双下肢活动基本正常,故被称为FAS[2],Katz则称之为“双上臂肌萎缩症”[3]。目前FAS的诊断主要依据其特征性的临床表现,但是FAS患者的症状个体差别较大,有研究显示,23%的FAS患者仅表现为下运动神经元受损的体征,而77%则呈现上运动神经元损害的症状与体征[2],延髓和下肢受累轻微[4]。尽管部分患者仅表现为下运动神经元损害而与进行性脊髓性肌萎缩(progressive muscular atrophy,PMA)有一定的重叠,但是由于其具有相对良性的进程[3,5]和临床症状分布的特殊性,目前的研究者还是倾向于FAS可能是ALS的一种良性的变异类型[2]。
本文8例患者以双上肢下运动神经元受累为突出表现,多数患者出现锥体束征,肌电图提示神经源性损害,根据Escorial诊断标准,符合ALS诊断。依据其双上肢对称性肌无力和肌萎缩等临床特征,考虑FAS诊断。FAS的肌肉病理改变与ALS相一致,更进一步印证其为ALS变异型的观点。而其临床表现、影像学、实验室及电生理检查均可排除颈椎病、双侧臂丛病变、糖尿病性周围神经病变、肯尼迪病、肌病、平山病等。
一般认为FAS患者有相对于ALS较长的生存期,呼吸功能衰竭多出现在病程晚期,自病程起始到使用呼吸机的平均时间为51个月[6],而ALS患者最小生存期则为20个月[7]。但是,Kataoka等[8]报道了5例FAS患者在早期出现呼吸衰竭,自病程开始到出现呼吸衰竭的平均时间为14个月,3例患者在出现呼吸衰竭后1个月内死亡。另有文献报道,FAS患者在未出现延髓麻痹的情况下,于病程早期发生了呼吸肌麻痹[9]。所以,FAS是否有相对于ALS良性的病程,还有待进一步观察研究。至少不能把FAS看做是ALS良性类型的决定性因素。
目前FAS的病因机制不明。国内外的病例报道中基本以男性患者为主,男女比例为9∶1,本文男女比例为7∶1,均明显高于ALS的1.5∶1,说明该病发病机制和经典的ALS不同。不同种族之间亦存在差异,黑人较白人的预后差[10]。以上均提示遗传学因素在发病中起着重要作用。这也是今后研究的方向之一。
1 Andersen PM,Borasio GD,Dengler R,et al.Good practice in the management of amyotrophic lateral sclerosis:clinical guidelines.An evidence-based review with good practice points.EALSC Working Group[J].Amyotroph Lateral Scler,2007,8(4):195-213.
2 Hu MT,Ellis CM,Al-chalabi A,et al.Flail arm syndrome:a distinctive variant of amyotrophic lateral sclerosis[J].J Neurol Neurosurg Psychiatry,1998,65(6):950-951.
3 Katz JS,Wolfe GI,Andersson PB,et al.Brachial amyotrophic diplegia:a slowly progressive motor neuron disorder[J].Neurology,1999,53(5):1071-1076.
4 Couratier P,Truong C,Khalil M,et al.Clinical features of flail arm syndrome[J].Muscle Nerve,2000,23(4):646-648.
5 Turner MR,Parton MJ,Shaw CE,et al.Prolonged survivial in motor neuron disease:a descriptive study of the king's database 1990—2002[J].J Neurol Neurosurg Psychiatry,2003,74(7):995-997.
6 Wijesekera LC,Mathers S,Talman P,et al.Natural history and clinical features of the flail arm and flail leg ALS variants[J].Neurology,2009,72(12):1087-1094.
7 Chiò A,Logroscino G,Hardiman O,et al.Prognostic factors in ALS:a critical review[J].Amyotroph Lateral Scler,2009,10(5/6):310-323.
8 Kataoka H,Kiriyama T,Kitauti T,et al.Flail arm syndrome with motor neuron disease rapidly progressing to respiratory failure:a case series and clinical analysis[J].Eur J Neurol,2010,17(9):e90-e91.
9 Ramdass R,Lekwuwa GU,Mitchell D.Diaphragmatic involvement without bulbar dysfunction or features to suggest amyotrophic lateral sclerosis in brachial amyotrophic diplegia[J].Muscle Nerve,2009,40(6):1066-1067.
10 Tomik B,Nicorta A,Ellis CM,et al.Phenotypic dierences between African and white patients with motor neuron disease:a case-control study[J].J Neurol Neurosurg Psychiatry,2000,69(2):251-253.
Clinical Analysis of Flail Arm Syndrome
WANG Te,SUN Xin-gang,YANG Huan,et al.Department of Neurology,Xiangya Hospital of Central-south University,Changsha 410008,China
ObjectiveTo study the clinical features of flail arm syndrome(FAS).MethodsClinical data of 8 cases of FAS admitted to the department of neurology in our hospital from June 1st 2010 to December 1st 2011 were retrospectively analyzed to summarize its clinical features.ResultsThe 8 cases in the study were mainly characterized by muscle atrophy of proximal upper extremity,mostly in middle-aged people〔(48.2±4.8)years〕and males(the ratio of male to female was 7∶1).The initial symptoms were mainly weakness of bilateral upper extremities(62.5%),accompanied by fasciculation.EMG showed extensive neurogenic damage(three or more than three segments).Muscle biopsy showed 8 cases had muscle fiber atrophy,7 cases had type grouping and 3 cases had target fibers.ConclusionFAS patients have a relatively slow progression of disease and longer survival time.It might be a clinical variant of typical amyotrophic lateral sclerosis and its morbidity might be related to genetic abnormality and race.
Flail arm syndrome;Amyotrophic lateral sclerosis;Motor neuron disease
R 746.4
B
1007-9572(2012)11-3776-03
10.3969/j.issn.1007-9572.2012.11.068
410008湖南省长沙市,中南大学湘雅医院神经内科
刘运海,410008湖南省长沙市,中南大学湘雅医院神经内科;E-mail:wangteson@hotmail.com
2012-05-13;
2012-10-09)
(本文编辑:张小龙)
猜你喜欢
保健与生活(2022年13期)2022-07-06
农村百事通(2022年16期)2022-07-06
农村百事通(2022年6期)2022-06-21
考试与评价·高二版(2020年2期)2020-09-10
武警医学(2019年2期)2019-03-05
火花(2018年2期)2018-11-21
幸福家庭(2018年11期)2018-11-13
体育科技文献通报(2017年4期)2017-11-27
中西医结合心脑血管病杂志(2016年20期)2016-03-01
中国康复理论与实践(2015年7期)2015-05-09
