
2-(2-羟基苯乙烯)基-4,6-二甲基均三嗪分子内质子转移的理论研究
2011-12-27 03:50王华静周子彦于先进
东北师大学报(自然科学版) 2011年3期
王华静,周子彦,于先进
(山东理工大学化学工程学院,山东淄博 255049)
2-(2-羟基苯乙烯)基-4,6-二甲基均三嗪分子内质子转移的理论研究
王华静,周子彦,于先进
(山东理工大学化学工程学院,山东淄博 255049)
利用量子化学的DFT/B3LYP方法,在6-311+G*水平上,研究了2-(2-羟基苯乙烯)基-4,6-二甲基均三嗪分子醇式和酮式结构互变异构化反应.对反应势能面的研究发现,标题化合物至少有8种异构体和8个过渡态,通过振动分析和内禀反应坐标分析对过渡态进行了研究.结果表明:所有醇式结构的异构体都比酮式结构的能量低;在室温下分子内质子转移由醇式向酮式转变难以进行,其转变最小的活化能为267.8 kJ/mol.
2-(2-羟基苯乙烯)基-4,6-二甲基均三嗪;质子转移;密度泛函理论;量化计算
1,3,5-均三嗪是重要的含氮六员杂环化合物,它们的同分异构体及衍生物广泛存在于生物体中,是决定生物体各种生物功能的天然化合物最关键的结构单元之一[1],是重要的有机化工原料和中间体,在制药、纺织、塑料等工业以及杀虫剂、染料、炸药、表面活性剂、有机光电功能材料等方面都有着广泛的应用[2-3],近年来,对均三嗪及其衍生物的理论和实验研究一直非常活跃[4-5].2-(2-羟基苯乙烯)基-4,6-二甲基均三嗪存在多种互变异构体(见图1),在这些异构体之间,可以通过化学键旋转和质子转移发生结构互变,质子转移产生的互变异构现象,不但在一般化学研究中会发生,而且在分子生物学上也同样发生,这对研究分子生物学具有非常重要的意义[6-8].为了进一步研究三嗪体系中的质子转移过程,本文用密度泛函理论的B3LYP/6-311+G*方法研究了2-(2-羟基苯乙烯)基-4,6-二甲基均三嗪质子转移的异构化过程[9-13],探讨了异构化反应的机理.
1 模型与方法
本文利用密度泛函理论的B3LYP方法,在6-311+G*基组下对2-(2-羟基苯乙烯)基-4,6-二甲基均三嗪(T1)异构化过程中的异构体和过渡态的几何构型进行了全优化.
在同一水平上进行了振动分析,确认了异构体的稳定态和一阶鞍点过渡态,通过内禀反应坐标分析对过渡态进行了进一步确认,反应过程中单点能进行了零点能校正.计算中所有的收敛精度均取程序设定的缺省值(10-8),全部计算在Internal Core2 1.63 GHz微机上用Gaussian 03量子化学程序包完成[14].
2 结果与讨论
根据图1所示的标题化合物结构以及产生的同分异构体,设计了可能的反应途径,首先利用B3LYP/6-311+G*方法对8个异构体(M1-M8)和其相应的过渡态构型进行全优化,在同一水平上进行了频率分析,所有的异构体的振动频率全部为正值,说明它们均可以稳定存在.而所有的过渡态的振动频率则有且只有一个虚振动频率,表明它们分别是各自势能面上的一级鞍点,通过内禀反应坐标分析表明过渡态两边所连接的是其对应的稳定异构体,表明过渡态是真实可信的.
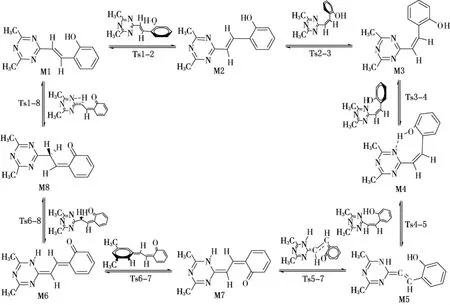
图1 分子内质子转移和旋转异构化反应路径
异构体和过渡态的稳定构型如图1所示,分子内质子转移和旋转异构化过程中各异构体和过渡态结构的零点能、总能量及振动频率见表1,反应途径及其能垒见图2.
化合物M1-M4都是醇式结构,其中M1和M2是反式结构,M3和M4是顺式结构,苯环与三嗪环之间的夹角分别为28.8°,26.5°,79.3°和22.9°.由于M4分子内存在1个H…N键长为0.165 8 nm,∠O—H…N为173.0°的氢键,比M3稳定,M4中苯环与三嗪环的夹角是最小的,两环的共轭程度最好;M6-M8是酮式结构,都是反式结构,苯环与三嗪环之间的夹角分别为0.03°,0.02°和0.45°,两环在同一个平面上;M5是双烯结构,苯环与三嗪环之间的夹角为88.4°,两环基本上是垂直的,══C C C之间的夹角为164.8°,基本上接近直线.
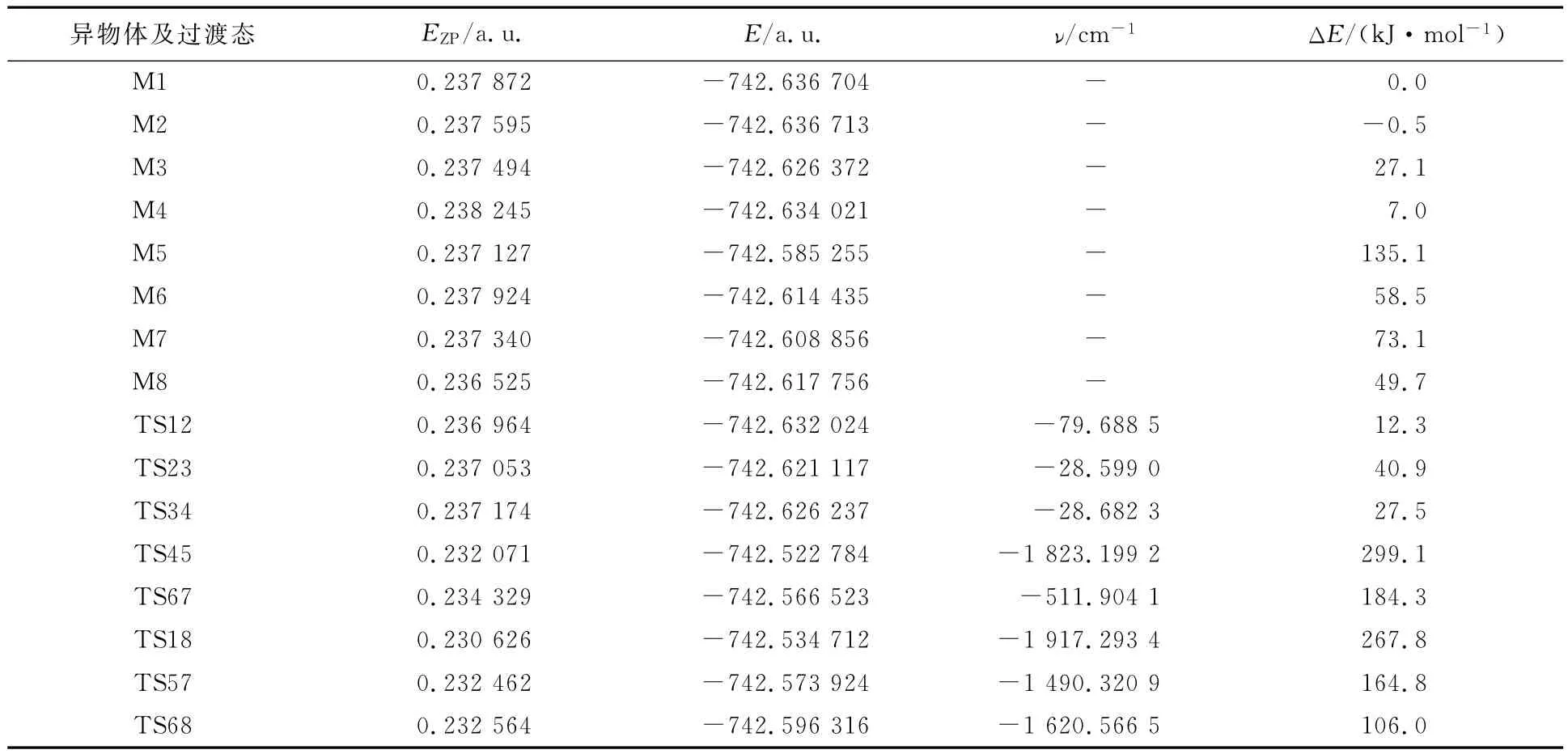
表1 分子内质子转移和旋转异构化反应路径中各异构体和过渡态的零点能EZP,总能量E及谐振频率
比较8种化合物的能量,M1-M8的能量大小顺序为:M5>M7>M6>M8>M3>M4>M1>M2.醇式结构的能量比酮式的低,其中醇式结构M2的能量是最低的,双烯式结构M5的能量最高,根据总能量越小越稳定的原理,可以得出M2构型是所有单体中最稳定的,M5是最不稳定的.从M1到M2进行异构化反应是放热反应,其热效应为0.5 kJ/mol,正、逆活化能分别为12.3和12.8 kJ/mol,室温下就能进行;而从M1到M8进行异构化反应却是吸热反应,其热效应为49.7 kJ/mol,正、逆活化能分别为267.8和218.1 kJ/mol,此反应是很难进行的;虽然M5转变成M7的活化能只有29.7 kJ/mol,但M4转变成M5的活化能却高达292.1 kJ/mol,也是难以进行的,因此通过分子内旋转和质子转移不能使醇式结构转变成酮式结构.根据阿累尼乌斯公式可知,在298K时,最稳定的结构M2与M1,M3,M4,M8存在的比例为1∶0.82∶0.049∶1.5×10-5∶1.6×10-9,说明常温下是以M2和M1的形式存在的.
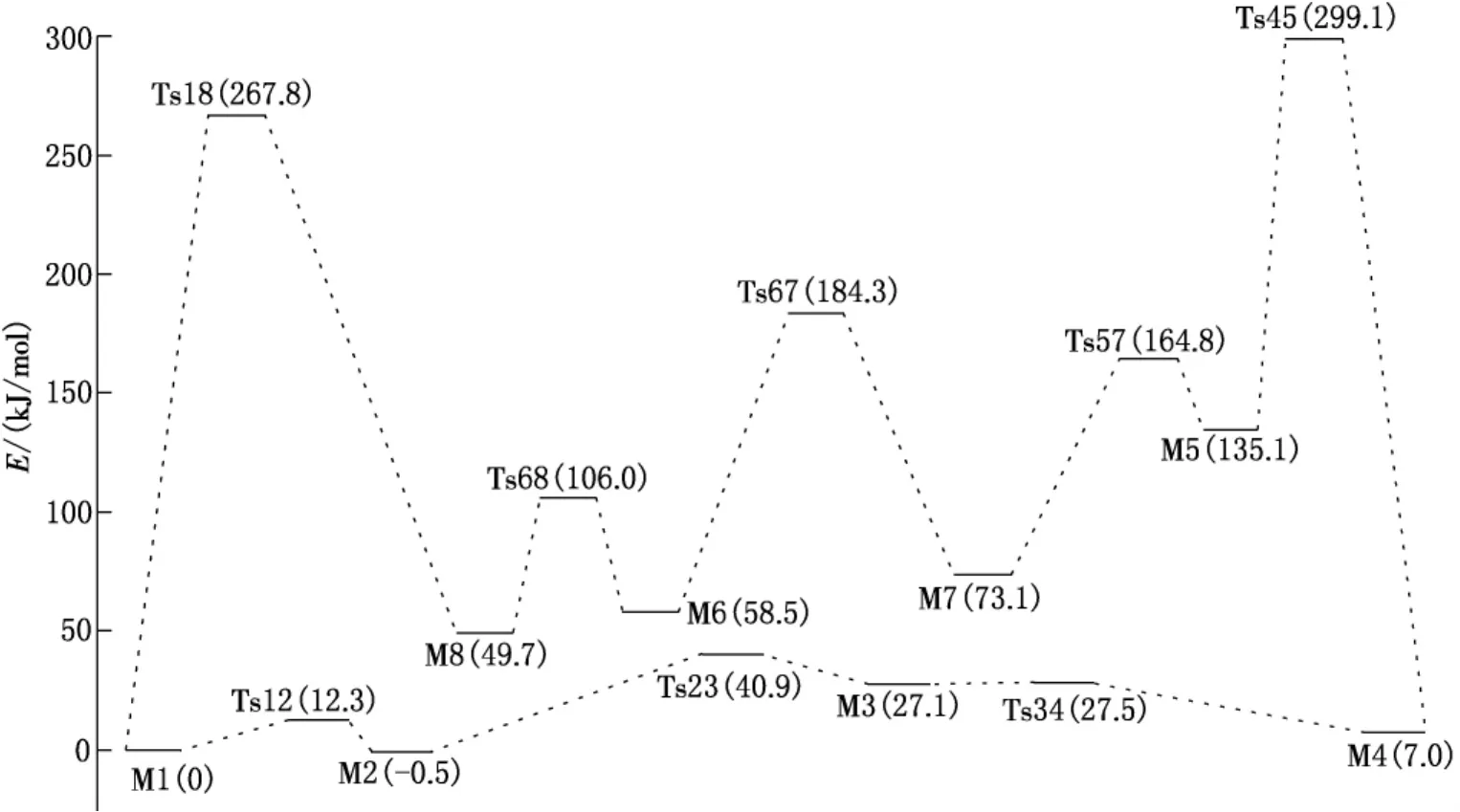
图2 T1基态分子内旋转和质子转移势能面图
图3为反应M5→Ts5-7→M7和M1→Ts1-8→M8异构化过程中经内禀反应坐标分析分子的关键部位的几何参数.
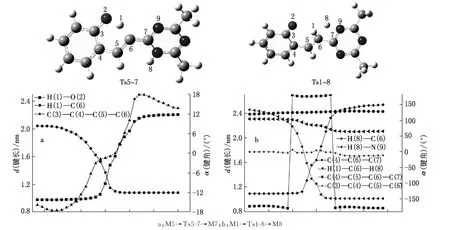
图3 M5→Ts5-7→M7和M1→Ts1-8→M8分子内异构化反应路径中关键部位的几何构型参数变化的内禀反应坐标分析
从图3(a)可以看出,在反应过程中,H(1)—O(2)键长增加,H(1)—C(6)键长减小,说明H(1)原子由O(2)原子向C(6)移动,发生分子内质子转移,∠C(3)—C(4)—C(5)—C(6)在反应过程中,由负值逐渐增大变为0,形成一个平面六元环过渡态,随着反应进一步进行,H(1)移至C(6),H(1)—C(6)键进一步缩短,∠C(3)—C(4)—C(5)—C(6)由0变成正值,形成产物M(7).从图3(b)可知,H(8)—C(6)键长增加,H(8)—N(9)键长在减小,H(8)原子从C(6)原子向N(2)移动,形成一个四元环过渡态,随着反应进一步进行,H(8)移至N(9),H(8)—N(9)键进一步缩短,最后成键形成产物M(8).C(5)—C(6)—C(7)和H(8)—C(6)—H(1)变化不太大,∠C(3)—C(4)—C(5)—C(6)变化也较小,只有∠C(4)—C(5)—C(6)—C(7)从-170°变化到170°后又变化到-170°,进而达到稳定构型,由于四元环的张力比六元环的大,所以过渡态Ts1-8的能量比过渡态Ts5-7的高.
3 结论
(1)计算结果表明,醇式结构异构体比酮式的稳定,常温下是以醇式结构稳定存在的.
(2)醇式结构异构体之间互相转化时所需的活化能较低,常温下就能进行.
(3)醇式结构异构体转化成酮式结构异构体时所需的活化能较高,常温下是很难进行的.
[1]花文廷.杂环化学[M].北京:北京大学出版社,1991:482.
[2]GRZEGORZ B.Recent applications of 2,4,6-trichloro-1,3,5-triazine and its derivatives in organic synthesis[J].Tetrahedron,2006,62:9507-9522.
[3]SUN Y,ZHAO D,HAROLD S F.Synthesis and properties of disperse dyes containing a built in triazine stabilizer[J].Dyes and Pigment,2007,74:608-614.
[4]崔月芝,方奇,薛刚,等.多支结构的均三嗪衍生物的合成及双光子吸收性质[J].化学学报,2005,63:1421-1428.
[5]ZHU W H,WU G S.An ab initio study of the first hyperpolarizabilities of octupolar substituted triazines:electron correlation,solvent effect and frequency dispersion[J].Chem Phys Lett,2002,358:1-7.
[6]袁永梅.5-甲基-3-硝基-4-异唑甲酰腙的晶体结构、电子光谱、抑菌活性及其量化计算[J].分子科学学报,2010,26(5):347-352.
[7]ZHOU Z Y,SHAN G G,LIAO Y,et al.A theoretical study of ground and excited state proton transfer and rotamerim in salicylanilide and its 1∶1 complex with methanol[J].J Mol Struct Theochem,2010,945:110-115.
[8]ZHOU Z Y,SHAO G G,ZHU Y L,et al.Theoretical studies on excited proton transfer tautomerism reaction and spectroscopic properties of 8-hydroxyqunoline monomers and dimers[J].J Struct Chem,2009,50(4):606-612.
[9]陈红,王春忠,王登攀,等.Co掺杂对单斜LiMnO2结构与性能的影响[J].吉林大学学报:理学版,2010,48(3):473-477.
[10]王立敏,刘丽,谢世伟,等.双阴离子吗啉型离子液体结构性能的量子化学研究[J].东北师大学报:自然科学版,2009,41(3):89-93.
[11]杨静,曾余瑶,徐文国,等.甲醛与臭氧反应机理的理论研究[J].东北师大学报:自然科学版,2010,42(1):89-92.
[12]钟爱国.双核镉配聚物及其衍生物荧光光谱的密度泛函理论研究[J].分子科学学报,2009,25(5):357-360.
[13]罗洪娟,黎安勇.B3N3H6与HF,HCl和H2O形成的氢键和双氢键[J].吉林大学学报:理学版,2009,47(2):376-382.
[14]FRISCH M J,TRUCKS G W,SCHLEGEL H B,et al.Gaussian 03.Revision B.03.Pittsburgh[DB].PA:Gaussian Inc,2003.
A theoretical study of ground state proton transfer rotamerim in 2-((2-hydroxy-styryl)yl)-4,6-dimethyl-1,3,5-triazine
WANG Hua-jing,ZHOU Zi-yan,YU Xian-jin
(College of Chemical Engineering,Shandong University of Technology,Zibo 255049,China)
Density functional theory(DFT)of quantum chemistry B3LYP method at the 6-311+G*basis set was used to investigate the enol form and keto form tautomerism reaction of 2-((2-hydroxystyryl)yl)-4,6-dimethyl-1,3,5-triazine which was mentioned in title.By the result of investigation of potential energy curves,it was found that there were 8 monomer isomers and 8 transition states.In addition,the transition states had been explored and proved by vibration analysis and intrinsic reaction coordination analysis.The results showed that energy of all the isomers of enol forms are lower than keto forms.The lowest reaction barrier of the proton transfer between the enol and keto form was 267.8 kJ/mol,which showed it was quite difficult to occur in room temperature.
2-((2-hydroxy-styryl)yl)-4,6-dimethyl-1,3,5-triazine;proton transfer;density functional theory;quantum chemistry calculation
O 641
150·30
A
1000-1832(2011)03-0093-04
2011-04-12
山东省自然科学基金资助项目(ZR2009BL024).
王华静(1986—),女,硕士研究生;周子彦(1962—),男,博士,教授,主要从事应用量子化学研究.
石绍庆)
猜你喜欢
系统仿真技术(2022年4期)2023-01-17
北京航空航天大学学报(2022年5期)2022-06-06
浙江化工(2022年4期)2022-05-07
石油石化绿色低碳(2019年6期)2019-01-14
电脑知识与技术(2018年3期)2018-03-21
哈尔滨理工大学学报(2017年1期)2017-04-08
科技视界(2016年24期)2016-10-11
国外医药(抗生素分册)(2016年4期)2016-07-12
信息记录材料(2016年4期)2016-03-11
化工进展(2015年3期)2015-11-11
