
β-淀粉样蛋白对BV2小胶质细胞促炎作用的研究
2011-12-15 07:18王爱桃熊永洁闫秋月张苏明
中国组织化学与细胞化学杂志 2011年5期
吴 军 王爱桃 闵 喆 熊永洁 闫秋月 张苏明*
β-淀粉样蛋白对BV2小胶质细胞促炎作用的研究
吴 军1,2王爱桃3闵 喆1熊永洁1闫秋月1张苏明1*
(1华中科技大学同济医学院附属同济医院神经内科,武汉430030;2郑州大学第一附属医院神经内科,郑州450052;3华中科技大学同济医学院附属协和医院麻醉科;武汉430022)
目的 探讨β-淀粉样蛋白(β-amyloid,Aβ)促进BV2小胶质细胞产生炎性因子IL-1β和TNFα的作用机制。方法体外培养BV2小胶质细胞,应用Aβ1-42作用于BV2小胶质细胞,或用吡咯烷二硫代氨基甲酸盐(PDTC)预孵育再给予Aβ1-42刺激,实时荧光定量反转录聚合酶链反应法(RT–PCR)检测IL-1β和TNFαmRNA表达;免疫印迹法(Western blot)检测胞核中NF-κB p65及其抑制蛋白胞浆中IkBα的表达。结果 Aβ1-42作用于BV2小胶质细胞后,Westernblot显示胞浆内IkBα表达下降,胞核内NF-κB p65表达明显增加,RT-PCR测定IL-1β和TNFαmRNA的表达增加;给予NF-κB信号通路特异阻断剂PDTC后,胞浆IkBα的下降和胞核内NF-κB p65的增加均被抑制,同时IL-1β和TNFαmRNA的表达亦受到抑制,PDTC的抑制效果呈剂量依赖性。结论 Aβ可通过激活小胶质细胞NF-κB信号通路促进IL-1β和TNFα的表达。
炎症;β-淀粉样蛋白;白细胞介素1β;肿瘤坏死因子α;NF-κB
炎症存在于多种疾病发生发展的病理过程中,人们在研究阿尔茨海默病(Alzheimer’s disease,AD)时,发现在其发病的病理过程中炎症起着非常重要的作用[1-2],而β-淀粉样蛋白(Aβ)对于 AD的炎性反应起到举足轻重的作用:β-amyloid(Aβ)激活小胶质细胞产生诸如IL-1β和TNFα等炎性因子,这些炎症因子对包括神经元在内的各种细胞造成伤害,并引起其凋亡[3-5],而且这些炎症因子可以引起胶质细胞的进一步激活,导致恶性循环。本研究探讨了Aβ对BV2的致炎性及其对细胞内NF-κB信号通路的作用。
材料和方法
1.细胞及试剂
小鼠小胶质细胞系BV2cells购自中国医学科学院基础医学研究所细胞中心,NF-κB p65抗体购自Cell Signaling公司,IκBα抗体购自Abcam 公司,goat anti-rabbit IgG-PE 购自Santa Cruz公司,goatanti rabbit IgG (H+L)HRP购自 Thermo Fisher Scientific公司,Aβ1-42购自 Anaspec公司,将其溶于双蒸水,使其终浓度为0.1mmol/L,37℃孵化5d,使用时在培养液中将其稀释至5μmol/L。吡咯烷二硫代氨基甲酸盐(PDTC)购自Sigma公司,将其溶于双蒸水,使其终浓度为0.1mmol/L,使用时在培养 液 中 将 其 分 别 稀 释 至 2.5μmol/L、5μmol/L、10μmol/L和20μmol/L。
2.细胞培养和分组
加入含10%胎牛血清(FBS;GibcoBRL,Life Technologies,CA)、100U/ml青霉素和100U/ml链霉素的DMEM/F12培养液中,置于37℃,5%C02饱和湿度孵育箱内培养。实验分为两部分:第一部分:将培养细胞分成4组(每组重复6次):(1)对照组:仅用培养液;(2)PDTC组:在培养液中加入PDTC(5μmol/L);(3)Aβ组:在培养液中加入Aβ1-42(5μmol/L);(4)Aβ 加 PDTC 组:PDTC(5μmol/L)预处理30min后,给予 Aβ1-42(5μmol/L)孵育。第二部分:将培养细胞分成5组(每组重复6次):分别用 PDTC 0μmol/L、2.5μmol/L、5μmol/L、10μmol/L、20μmol/L 预处理30min后,再加入给予 Aβ1-425μmol/L孵育。
3.实时荧光定量PCR(RT-PCR)检测IL-1β和TNFαmRNA表达水平
用5μmol/L Aβ1-42刺激6h后,按说明书提取BV2细胞的总RNA,然后进行逆转录反应,反应体积25μl,其中:1μl总 RNA、1μl mol/L-MLV、4μl M-MLV RT 5 × buffer、20Uof Rnasin、1μl Random primer (10pmol/μl)、1μl of dNTP mix (10 mmol/L)和DEPC水。将其混合37℃孵育50min,70℃孵育15min,获得的cDNA在-20℃保存,PCR操作按LightCycler DNA的SYBR Green I的使用说明,经过1min、95℃的初始变性后,变性95℃、15s,复性58℃、15s,延伸72℃、45s,共40个循环,引物序列如下:
β-actin(正 义 链):5'-CCGTGAAAAGATGACCCAG-3',
β-actin(反义链):5'-TAGCCACGCTCGGTCAGG-3';
IL-1β(正义链):5'-TTTGAAGTTGACGGACCCC-3',
IL-1β (反 义 链 ) : 5' -GTGCTGCTGCGAGATTTGA-3';
TNFα (正 义 链 ) :5' -CGGGCAGGTCTACTTTGGAG-3',
TNFα (反 义 链):5'-CAGGTCACTGTCCCAGCATC-3'。
PCR数据分析采用Ct法。
4.免疫印迹法(Westem blot)检测 NF-κB p65和IkBα
用 Aβ1-42(5μmol/L)刺激4h后,裂解液提取胞浆蛋白和核蛋白,BCA法测定蛋白质浓度,蛋白上样量40μg,在SDS-聚丙烯酰胺凝胶中电泳,再转到NC膜上,室温下20%脱脂牛奶封闭1h,然后使用兔抗鼠多克隆抗体IκBα或 NF-κB p65或β-actin及LamineB14℃孵育过夜后,辣根过氧化物酶标记的羊抗兔IgG抗体室温孵育1.5h。加入ECL溶液1min,X光片曝光。
5.统计学方法
应用SPSS11.0统计分析软件进行统计学处理,实验结果以¯x±s表示,每个实验至少重复3次,组间比较采用单因素方差分析,P<0.05,有统计学意义。
结 果
1.RT-PCR
Aβ1-42处 理 BV2 细 胞 6h 后,IL-1βmRNA 和TNFαmRNA表达水平明显上调,给予NF-κB信号通路特异阻断剂PDTC预处理细胞30min后,再加Aβ1-42处理BV2细胞,则发现PDTC抑制 Aβ1-42所引起的IL-1β和TNFαmRNA上调(图1A,B),其抑制作用呈剂量依赖性(图1C,D)(▲P<0.05,*P<0.01)。
2.Western blot
Aβ1-42处理 BV2 细胞后4h后,胞浆内IκBα表达显著下降,胞核内NF-κB p65表达增加,给予NF-κB信号通路特异阻断剂PDTC预处理细胞30min后,再加Aβ1-42处理BV2细胞,则发现呈PDTC抑制 Aβ1-42诱导的IκBα降解和 NF-κB p65核内移(图2A,B),其抑制作用呈剂量依赖性(图2C,D)(*P<0.01)。
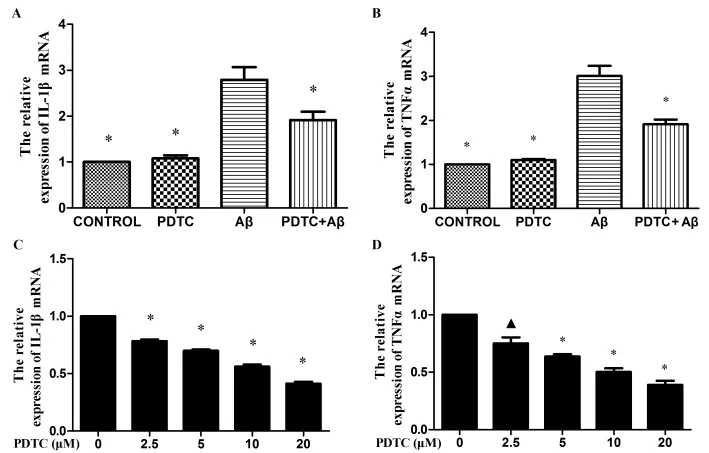
图1 RT-PCR 显示:Aβ1-42明显上调IL-1βm RNA 和 TNFαmRNA 水平,PDTC抑制 Aβ1-42所引起的IL-1βmRNA 和TNFαmRNA的上调(图1A,B),其抑制作用呈剂量依赖性(图1C,D)。Fig1RT-PCR assay for IL-1βand TNFαmRNA after BV2cells were treated with or without Aβ1-42(5μmol/L)in the absence or presence of PDTC (5μmol/L)for 6h.Aβ1-42markedly enhanced the gene expression of IL-1βand TNFα,PDTC alone did not affect gene expression.PDTC significantly attenuated the gene expressions of IL-1βand TNFαinduced by Aβ1-42(Fig 1A,B),and the effects were dose dependent(Fig1C,D),Data are presented as means±S.D.▲P<0.05,*P<0.01compared with Aβgroup.
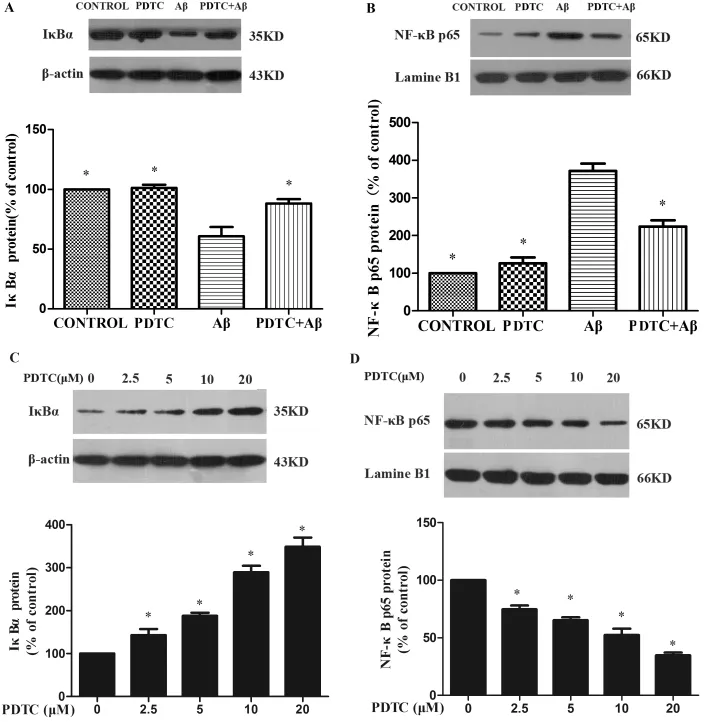
图2 2Western blot显示:Aβ1-42促进胞浆内IκBα表达显著下降、胞核内 NF-κB p65表达增加,PDTC抑制 Aβ诱导的IκBα降解和 NF-κB p65增加(图2A,B),其抑制作用呈剂量依赖性(图2C,D)。Fig2 Western blot analyses of IκBαand NF-κB p65after BV2cells were treated with or without Aβ1-42(5μmol/L)in the absence or presence of PDTC (5μmol/L)for 4h.Plasmic extracts or nuclear extracts were separated and blotted sequentially with the indicated antibodies to evaluate both IκBαprotein contents in cytoplasm and NF-κB p65protein contents in nucleus.Aβ1-42caused a marked degradation of IκBα,and then induced the NF-κB p65subunit to enter the nucleus,these reactions could be inhibited by PDTC (Fig 2A,B)and the inhibitory effects were dose dependent(Fig 2C,D).The data represent the mean±SD of at least three independent experiments.*P<0.01compared with Aβgroup.
讨 论
AD是一种中枢神经系统退行性疾病,主要表现为记忆和认知功能障碍、行为及人格异常改变。随着人口老龄化进程的加快,AD将成为威胁老年人的生活质量并导致死亡的最严重疾病之一。AD具有神经元丢失、淀粉样蛋白斑块和神经纤维缠结形成及脑血管淀粉样变性等特征性病理改变,但其发病机制至今仍不是十分清楚,越来越多来自人类和动物模型的证据显示炎症在AD发病机制中扮演着非常重要的作用[6],一些研究认为AD的病理改变就是炎症累积损伤的结果,并且在炎症的发展过程中,Aβ的神经毒作用举足轻重,Aβ能持续地激活炎症反应,并造成神经组织的损伤[6]。
随着研究的深入,逐渐发现Aβ可以激活胶质细胞特别是小胶质细胞[7]。小胶质细胞是中枢神经系统的免疫细胞,具有双向作用,既有对神经元的保护作用,也可以激活时产生大量炎性因子[8][9]对神经元起毒性作用[10],其中IL-1β促进tau蛋白磷酸化[11],TNFα促进 Aβ合成[12],这些最终增加了速淀粉样斑块和神经纤维缠结的形成,随着越来越多淀粉样斑块和神经纤维缠结形成及神经元受损,记忆和认知功能逐渐减退。因此以小胶质细胞及其产生的炎症因子作为研究的切入点对AD的防治具有现实意义。
本研究选用BV2细胞系为携带癌基因v-raf/vmyc的反转录病毒J2感染原代培养的小鼠小胶质细胞而获得的永生细胞系,具基本备原代培养的各项特点,且高度纯化,为研究Aβ对小胶质细胞作用的理想的模型。目前关于对Aβ对小胶质细胞作用的研究较少,对其作用机制的探讨更少,因此本实验着重关注Aβ对BV2细胞系的作用和可能的机制,这将为深入了解AD炎性反应及抗炎治疗提供有益的帮助和借鉴。
本研究离体给予Aβ作用于BV2小胶质细胞后,western blot显示胞浆内IkBα表达下降,胞核内 NF-κB p65表达明显增加,RT-PCR 测定IL-1β mRNA和TNFαmRNA的表达增加;给予NF-κB信号通路特异阻断剂PDTC后,胞浆IkBα的下降和胞核内NF-κB p65的增加均被抑制,同时还抑制了IL-1β和TNFαmRNA表达的增加,并且,PDTC的抑制作用呈剂量依赖性:随着PDTC剂量的增加,抑制作用逐渐增强。研究提示,Aβ作用于BV2小胶质细胞可能通过激活 NF-κB信号通路,促进IL-1β和TNFα等炎性因子的表达。
NF-κB信号通路常常在炎症过程中发挥关键作用,正常情况下,IkBα与有转录活性的二聚体是p50/p65(即通常所指的 NF-κB)结合形成三聚体,阻止细胞质中的NF-κB进入细胞核,Aβ刺激BV2细胞后,引起胞浆内IkBα降解、NF-κB p65核内移并与炎性因子的DNA结合,启动了其随后的转录调控功能,导致相应的炎性因子表达增加。有研究显示,作为AD标志性病理改变的细胞内神经纤维缠结和细胞外Aβ沉积均与NF-κB的激活有关[13]。这意味着抑制NF-κB的激活,不仅明显减轻Aβ所致的炎症反应,减少了炎性因子对神经元细胞的损害,同时还将减少细胞内神经纤维缠结和细胞外Aβ沉积,因此,通过抑制NF-κB激活,进而抑制炎症应作为AD抗炎治疗药物开发关注的靶点将具有重要临床意义。
[1]Hoozemans JJ,Veerhuis R,Rozemuller JM,et al.Neuroinflammation and regeneration in the early stages of alzheimer's disease pathology.Int J Dev Neurosci,2006,24(2-3):157-165
[2]Eikelenboom P,Veerhuis R,Scheper W,et al.The significance of neuroinflammation in understanding alzheimer's disease.J Neural Transm,2006,113(11):1685-1695
[3]Graeber MB,Streit WJ.Microglia:Biology and pathology.Acta Neuropathol,2010,119(1):89-105
[4]Moore AH,O'Banion MK.Neuroinflammation and antiinflammatory therapy for alzheimer's disease.Adv Drug Deliv Rev,2002,54(12):1627-1656
[5]Wang MJ,Lin WW,Chen HL,et al.Silymarin protects dopaminergic neurons against lipopolysaccharide-induced neurotoxicity by inhibiting microglia activation.Eur J Neurosci,2002,16(11):2103-2112
[6]Wyss-Coray T.Inflammation in alzheimer disease:Driving force,bystander or beneficial response?Nat Med,2006,12(9):1005-1015
[7]Rogers J,Lue LF.Microglial chemotaxis,activation,and phagocytosis of amyloid beta-peptide as linked phe-nomena in alzheimer's disease.Neurochem Int,2001,39(5-6):333-340
[8]Hanisch UK.Microglia as a source and target of cytokines.Glia,2002,40(2):140-155
[9]Minghetti L,Levi G.Microglia as effector cells in brain damage and repair:Focus on prostanoids and nitric oxide.Prog Neurobiol,1998,54(1):99-125
[10]Combs CK,Karlo JC,Kao SC,et al.Beta-amyloid stimulation of microglia and monocytes results in tnfalpha-dependent expression of inducible nitric oxide synthase and neuronal apoptosis.J Neurosci,2001,21(4):1179-1188
[11]Sheng JG,Zhu SG,Jones RA,et al.Interleukin-1 promotes expression and phosphorylation of neurofilament and tau proteins in vivo.Exp Neurol,2000,163(2):388-391
[12]Blasko I,Marx F,Steiner E,et al.Tnfalpha plus ifngamma induce the production of alzheimer beta-amyloid peptides and decrease the secretion of apps.FASEB J,1999,13(1):63-68
[13]Yamamoto Y,Gaynor RB.Role of the nf-kappab pathway in the pathogenesis of human disease states.Curr Mol Med,2001,1(3):287-296
Study on the inflammation induced byβ-amyloid in Bv2microglia cells
Wu Jun1,2,Wang Aitao3,Min Zhe1,Xiong Yongjie1,Yan Qiuyue1,Zhang Suming1*
(1Department of Neurology,Tongji Hospital,Tongji Medical College,Huazhong University of Science and Technology,Wuhan 430030;2Department of Neurology,The First Affiliated Hospital of Zhengzhou University,Zhengzhou450052;3Department of Anesthesiology,Union Hospital,Tongji Medical College,Huazhong University of Science and Technology,Wuhan 430022,China)
ObjectiveExplore the molecular mechanisms of the inflammation induced byβ-amyloid(Aβ)in BV2cells,a mouse microglial cell line.MethodsCultured BV2cells were treated with Aβ1-42with or without Pyrrolidinedithiocarbamate ammonium (PDTC).Then we used RT-PCR assay for interleukin-1β(IL-1β)and tumor necrosis factorα(TNFα)mRNA,western blot analyses for IκBαand NF-κB p65.ResultsPDTC attenuated the gene expressions of IL-1βand TNFα,inhibited the degradation of IκBαand the translocation of NF-κB p65subunits into the nucleus of Aβ-stimulated BV2cells,and the inhibitory effects were dose dependently elevated.ConclusionOur findings suggest that IL-1βand TNFαmRNA are induced by Aβvia the NF-κB signal pathway in BV2microglial cells.
Inflammation;β-amyloid;IL-1β;TNFα;NF-κB
R329
A
10.3870/zgzzhx.2011.05.001
2011-03-14
2011-05-19
国家“863”高技术研究发展计划资助(2007AA03Z312)
吴军,男 (1968年),汉族,副主任医师。
*通讯作者(To whom correspondence should be addressed)
猜你喜欢
神经损伤与功能重建(2020年10期)2020-12-23
神经损伤与功能重建(2020年11期)2020-12-01
江西医药(2020年4期)2020-04-28
生殖医学杂志(2019年6期)2019-06-24
神经损伤与功能重建(2018年2期)2018-02-01
中成药(2017年9期)2017-12-19
中成药(2017年5期)2017-06-13
标记免疫分析与临床(2016年9期)2016-11-21
中华老年多器官疾病杂志(2016年8期)2016-05-14
中国男科学杂志(2016年9期)2016-03-20
