
合成3-苯甲酰胺基-6-甲基-2-吡喃酮衍生物的新方法
2011-11-23 05:51潘成学黄映飞苏桂发
合成化学 2011年6期
潘成学, 黄映飞, 苏桂发
(广西师范大学 药用资源化学与药物分子工程国家重点实验室培育基地,广西 桂林 541004)
2-吡喃酮衍生物不仅具有非常重要的生理活性[1,2],而且还是非常重要的有机合成中间体[3],例如1和2(Chart 1)。 Hren等[4]将1和2分别与芳基脒或胍的衍生物反应合成了各种嘧啶化合物;Kranjc等将1和2与炔类化合物反应合成了多取代的芳胺[5]和吲哚[6], 或者与丁二酰亚胺衍生物反应合成多环化合物[7];Vranicar等[8]则用1和2与肼反应来合成氨基酸衍生物;Pozgan等[9]以1和2为原料来合成新的吡喃酮衍生物等。
鉴于1和2在杂环化学中的重要作用,其合成也倍受关注[10~12]。Svete等[10]以马尿酸(3),N,N-二甲基甲酰胺的二甲缩醛(DMFDMA)为原料,先合成出两者的缩合物,再与乙酰丙酮反应得到2。该方法虽然第二步的产率达73%,但3与DMFDMA的缩合产率只有17%,而且纯化很困难;同年Kepe等[11]将此合成方法改为一步法合成,分别以42%和23%的产率合成了1和2。鉴于DMFDMA价格比较贵,他们又用价格比较低廉的原甲酸三酯代替DMFDMA,但1(21%)和2(16%)的产率较低。

Chart1

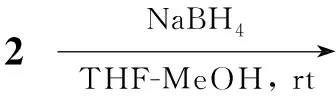
Scheme1
2005年,Anwar等[12]报道以3和原甲酸三乙酯为原料,先合成出噁唑酮4; 4经纯化后再与乙酰乙酸乙酯或乙酰丙酮在氢化钠存在下于二氧六环中回流,以50%左右的总产率得到1或2。该方法虽然总产率较高,但不仅要用强碱氢化钠,二氧六环必须经过严格的无水处理,而且对中间体4的纯度要求较高。
本文对文献[12]方法的合成路线进行了改进。以3,原甲酸三乙酯为原料,先制备4[13]; 4不经纯化,直接与过量的乙酰乙酸乙酯或乙酰丙酮在三乙胺的催化下加热反应;或者以二氯甲烷为溶剂,在三乙胺的催化下与乙酰乙酸乙酯或乙酰丙酮回流反应,以较高的总产率合成了1(66%)或2(60%); 2用NaBH4还原合成了一个新的吡喃酮衍生物5(Scheme 1)。
改进后的新工艺不仅对中间体4不用纯化,而且产物1和2也只需经过简单的重结晶即可得到纯品,具有操作非常简单,反应条件温和,总产率高等优点。
1 实验部分
1.1 仪器与试剂
X-4型数字显示显微熔点仪;Bruker Avance 500型超导核磁共振仪(CDCl3为溶剂,TMS为内标)。
所用试剂均为化学纯或分析析,其中乙酸酐、乙酰乙酸乙酯和乙酰丙酮使用前经重蒸纯化。
1.2 合成
(1) 4的合成[13]
搅拌下向反应瓶中依次加入3 5.4 g(30 mmol),乙酸酐6.5 mL(69 mmol),原甲酸三乙酯6.0 mL(36 mmol)及DMAP 约5 mg, N2保护下于110 ℃(浴温)回流反应1 h。减压蒸除溶剂得深红棕色油状4粗品(直接进行下步反应)。
(2)1和2的合成
方法一: 在4中加入乙酰乙酸乙酯或乙酰丙酮15 mL,搅拌下加入三乙胺1.5 mL, N2保护下于100 ℃(浴温)反应1.5 h。减压除去过量的1,3-二羰基化合物,冷却得固体,用混合溶剂A[V(二氯甲烷) ∶V(石油醚)=1 ∶2]溶解,于室温自然挥发,析出1或2。
方法二: 在4中加入CH2Cl250 mL,搅拌下加入乙酰乙酸乙酯或乙酰丙酮2 mL及三乙胺1 mL, N2保护下于65 ℃(浴温)回流反应2 h。减压除去CH2Cl2后用混合溶剂A重结晶得1或2。方法一和方法二的产率相近。
1: 淡紫色固体5.96 g,总产率66%, m.p.135 ℃~137 ℃;1H NMRδ: 8.83(s, 1H, NH), 8.54(s, 1H, ArH), 7.86~7.88(m, 2H, PhH), 7.47~7.57(m, 3H, PhH), 4.31~4.35(m, 2H, OCH2), 2.65(s, 3H, CH3), 1.36~1.39(m, 3H, CH3in Et);13C NMRδ: 165.93, 163.92, 162.83, 158.90, 133.40, 132.53, 128.92, 127.11, 124.03, 122.32, 109.98, 61.57, 19.56, 14.25。
2: 淡灰色固体4.88 g,总产率60%, m.p.130 ℃~134 ℃;1H NMRδ: 8.87(s, 1H, NH), 8.59(s, 1H, ArH), 7.86~7.88(m, 2H, PhH), 7.48~7.58(m, 3H, PhH), 2.59(s, 3H, CH3), 2.52(s, 3H, CH3);13C NMRδ: 195.95, 166.20, 161.91, 158.51, 133.23, 132.68, 129.00, 127.09, 123.50, 122.52, 116.57, 29.33, 19.94。
(3)5的合成
在反应瓶中加入2 1.36 g(5 mmol), THF 20 mL和甲醇2 mL,搅拌使其溶解;分批加入NaBH40.19 g(5 mmol),加毕,于室温反应30 min。加入乙酸乙酯50 mL;慢慢加水20 mL,搅拌2 min后静置。分液,有机层用饱和食盐水(20 mL)洗涤,无水Na2SO4干燥,减压除去溶剂后经硅胶柱层析[洗脱剂:V(石油醚) ∶V(乙酸乙酯)=3 ∶1]纯化或用混合溶剂[V(二氯甲烷) ∶V(石油醚)=1 ∶2]重结晶得淡黄色固体5 0.96 g,产率70%, m.p.153 ℃~154 ℃;1H NMRδ: 8.62(s, 1H, NH), 8.56(s, 1H, ArH), 7.83~7.85(m, 2H, PhH), 7.46~7.57(m, 3H, PhH), 4.84(d,J=4.5 Hz, 1H, CH), 2.23(s, 3H, CH3), 1.44~1.45(d,J=4.5 Hz, 3H, CH3);13C NMRδ: 166.10, 159.97, 150.81, 133.45, 132.49, 128.90, 127.11, 124.05, 123.34, 120.23, 64.78, 23.25, 16.40。
2 结果与讨论
我们曾按文献[11,12]方法来合成1和2,由于产率较低,产品的纯化很麻烦;而如果把底物的量由文献[11]报道的4 mmol扩大到20 mmol,产率则急剧下降,基本上不能分离出产物。文献[12]方法虽然产率较高(4→1或2的产率为80%),但对4的纯度要求较高。
经过实验研究发现,如果用三乙胺作为碱,利用过量的乙酰乙酸乙酯或乙酰丙酮与纯度较高的4反应,4的转化基本上是定量的;而粗品4直接与过量的乙酰乙酸乙酯或乙酰丙酮反应合成1或2,则两步总产率高达66%。
在2经硼氢化钠还原合成5的反应中,我们先尝试在甲醇中反应,但产物比较复杂,经过不断摸索,发现用混合溶剂[V(甲醇) ∶V(THF)=1 ∶10]效果最好,TLC检测发现反应几乎只有一个新点生成;随着反应溶剂中甲醇用量的进一步降低,会导致反应时间延长、底物反应不完全;溶剂中甲醇的量增多则反应产率降低,这可能是增加甲醇的量,会导致原料或产物发生开环。
[1] Sunazuka T, Omura S. Total synthesis ofα-pyrone meroterpenoids,novel bioactive microbial metabolites[J].Chem Rev,2005,105(12):4559-4580.
[2] Mcglacken G P, Fairlamb I J S. 2-Pyrone natural products and mimetics:Isolation,characterisation and biological activity[J].Nat Prod Rep,2005,22(3):369-385.
[3] 周庆发,赵慎,王珣,等. 2-吡喃酮衍生物合成研究进展[J].有机化学,2010,30(11):1652-1663.
[4] Hren J, Pozgan F, Buuic A,etal. An expeditious synthesis ofβ-pyrimidyl-α,β-didehydro-α-amino acid derivatives and pyrano[2,3-d]pyrimidines using microwave-assisted conditions[J].Tetrahedron,2009,65(39):8216-8221.
[5] Kranjk K, Kocevar M. Ethyl vinyl ether as a synthetic equivalent of acetylene in a DABCO-catalyzed microwave-assisted Diels-Alder-elimination reaction sequence starting from 2H-pyran-2-ones[J].Synlett,2008,(17):2613-2616.
[6] Kranjk K, Kocevar M. An expedient route to indoles via a cycloaddition/cyclization sequence from (Z)-1-methoxybut-1-en-3-yne and 2H-pyran-2-ones[J].Tetrahedron,2008,64(1):45-52.
[7] Kranjk K, Kocevar M. Microwave-assisted Diels-Alder reaction of 2H-pyran-2-ones with maleimides towards fused bicyclo[2.2.2]octenes[J].Heterocycles,2007,73:481-491.
[8] Vranicar L, Pozgn F, Polanc S,etal. Synthesis and transformations of a pyrazole containingα,β-didehydro-α-amino acid derivatives[J].Amino Acids,2003,24(3):273-280.
[9] Pozgan F, Krejan M, Polanc S,etal. 5-Acyl-2H-pyran-2-ones in the Schmidt reaction:Migration of the pyran-2-one ring[J].Heterocycles,2006,69:123-132.
[10] Svete J, Cadez Z, Stanovnik B,etal. Methyl 2-(benzoylamino)-3-(dimethylamino)-propenoate in the synthesis of heterocyclic systems.The synthesis of substituted 3-benzoylamino-2H-pyran-2-ones[J].Synthesis,1990,(1):70-72.
[11] Kepe V, Kocevar M, Polanc S,etal. A simple and general one-pot synthesis of some 2H-pyran-2-ones and fused pyran-2-ones[J].Tetrahedron,1990,46(6):2081-2088.
[12] Anwar H F, Metwlly N H, Gaber H,etal. The behavior of 4-alkoxymethylene-2-phenyl-4H-oxazol-5-one and 4-dimethylaminomethylene-2-phenyl-4H-oxazol-5-one toward nitrogen nucleophiles under microwave heating[J].J Chem Res,2005,(1):29-31.
[13] Matos M R, Gois P M, Mata M L,etal. Studies on the preparation of 4-ethoxyalkyliden and 4-aminoalkyliden-5(4H)-oxazolones[J].Syn Commun,2003,33(8):1285-1299.
猜你喜欢
应用化工(2023年1期)2023-02-16
今日农业(2022年13期)2022-11-10
化学工程师(2022年5期)2022-05-11
今日农业(2021年15期)2021-11-26
河南化工(2020年11期)2020-12-10
今日农业(2020年17期)2020-10-27
浙江化工(2018年3期)2018-04-19
合成化学(2015年9期)2016-01-17
微生物学杂志(2015年1期)2015-12-26
烟草科技(2015年8期)2015-12-20
