
大粒径纤维素衍生物手性固定相合成及对普萘洛尔的手性拆分研究
2011-08-06 07:35杨沛王荣马骏谢华张军莉贾正平武晓玉王娟兰州军区兰州总医院药理基地兰州市730050兰州大学药学院兰州市730000
中国药房 2011年33期
杨沛,王荣,马骏,谢华,张军莉,贾正平#,武晓玉,王娟(.兰州军区兰州总医院药理基地,兰州市 730050;2.兰州大学药学院,兰州市 730000)
随着对映体化学研究的不断深入,人们越来越认识到手性化合物在医药、农药及其他生物功能材料方面的重要作用[1]。对映异构体的手性拆分是获得光学纯物质的有效手段,手性高效液相色谱(Chiral HPLC)是近年来迅速发展起来的手性拆分方法,在药物、天然产物和不对称合成等研究领域应用前景广阔[2,3]。手性HPLC的核心是手性固定相,其中由Kubota等[4,5]发展起来的多糖衍生物类手性固定相,通用性强、经久耐用、样品载荷量大[6],被广泛地用于各种类型外消旋化合物的分析或制备拆分中[7]。
在多糖衍生物类手性固定相中,纤维素苯基氨基甲酸酯类衍生物特别是纤维素-三(3,5-二甲基苯基氨基甲酸酯)(CDMPC)对很多手性化合物都有极好的手性拆分能力[6]。Okamoto等[8]曾用CDMPC对510个消旋体进行手性拆分,其中229个完全拆分,86个部分拆分。虽然此类手性色谱柱已商品化,但商品化的手性色谱柱均采用小粒径硅胶(5、10µm)为基质,虽然柱效高、分离效果好,但柱压较高且价格昂贵,不适用于对映体的制备分离。而采用大粒径硅胶(40~60µm)作为填料基质,虽然柱效有所下降,但却克服了以小粒径硅胶作为填料基质成本高、柱压高的缺点,在手性制备柱的应用上显示了很好的前景。因此,本研究以分离制备手性对映异构体为研究目的,为降低运行成本,合成了大粒径涂敷型CDMPC手性固定相。并根据此类固定相适合于分离胺、醇、醚及其衍生物的性质[9],选择了结构中含有羟基、氨基及醚键的普萘洛尔作为评价对象,通过对普萘洛尔对映体的分离评价了合成的大粒径涂敷型CDMPC手性固定相的手性拆分性能,并探讨了大粒径手性固定相的拆分机制,为进一步开发低成本的分析及半制备手性固定相奠定基础。
普萘洛尔化学结构式见图1。
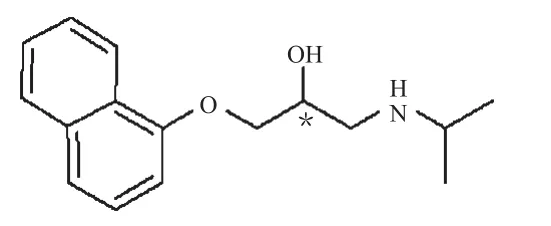
图1 普萘洛尔化学结构式Fig 1 Chemical structure of propranolol
1 仪器与试药
1.1 仪器
NEXUS 670傅里叶变换红外光谱仪(美国Nicolet公司);Vario EL元素分析仪(德国Elementar Analysensysteme GmbH公司);液相色谱系统,包括LC-6A泵、SPD-6AV紫外检测器(日本Shimadzu公司);分析之星色谱工作站(天津奥特赛斯仪器有限公司)。
1.2 试药
硅胶(粒径40~60 µm)、3-氨丙基三乙氧基硅烷、3,5-二甲基苯基异氰酸酯(美国Acros Organics公司);微晶纤维素(以下简称纤维素,上海恒信化学试剂有限公司);普萘洛尔标准品(中国药品生物制品检定所,批号:100783-200401,纯度:≥99.9%);其余试剂均为分析纯。
2 方法与结果
2.1 标准品溶液的制备
精密称取普萘洛尔标准品2 mg至5 mL容量瓶中,加甲醇至刻度,摇匀,即得。
2.2 手性色谱柱的制备
2.2.1 氨丙基烷化硅胶(APS)的合成。APS的合成参考文献[10],取硅胶50.08 g经盐酸活化后,置于1 000 mL三颈瓶中,180℃下真空干燥5 h,得活化硅胶。在N2保护下,加入500 mL无水苯(经CaH2干燥)和9 mL无水吡啶(经CaH2干燥),再加入3-氨丙基三乙氧基硅烷13 mL,80℃下回流反应24 h(电磁搅拌),冷却后,分别依次用适量的甲醇、丙酮、正己烷清洗,60℃下真空干燥5 h,产物置于干燥器中备用。合成反应见图2。
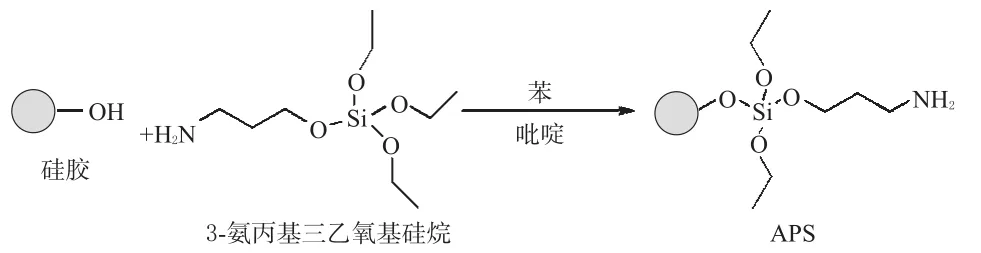
图2 APS合成示意图Fig 2 Synthesis diagram ofAPS
2.2.2 CDMPC的合成。参照文献[11]将3.05 g纤维素置于500 mL三颈瓶中,90℃下真空干燥5 h。在N2保护下,加入120 mL吡啶,加热回流24 h后,再加入30 mL 3,5-二甲基苯基异氰酸酯;100℃下油浴反应48 h(电磁搅拌),冷却至室温,剧烈搅拌下加入大量甲醇,抽滤,得灰白色固体,再用适量的甲醇洗涤后,60℃下真空干燥24 h,得灰白色固体9.73 g,产率85.78%。合成路线见图3。
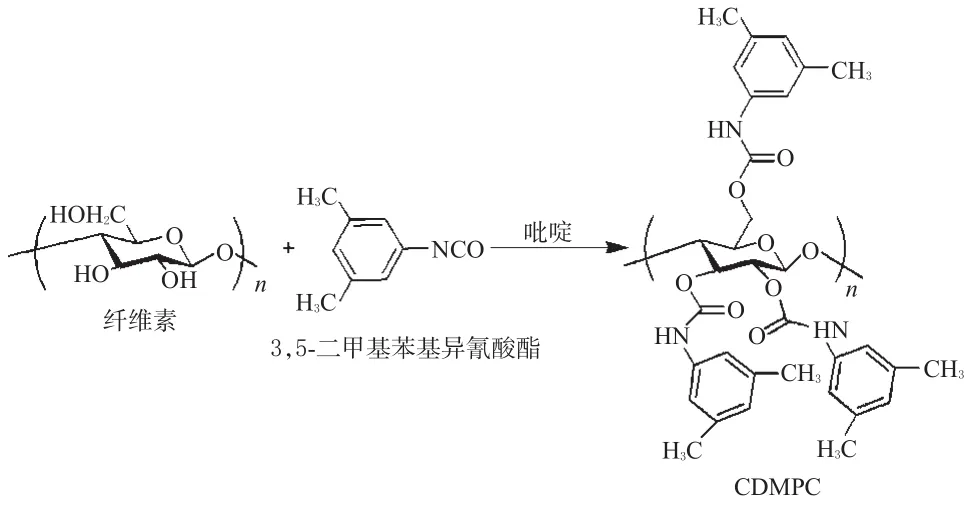
图3 CDMPC合成示意图Fig 3 Synthesis diagram of CDMPC
2.2.3 手性固定相的涂敷。取8.02 g CDMPC溶于400 mL四氢呋喃中,过夜后取出100 mL,加入到40 g APS中,磁力搅拌20 min使其分散均匀,真空旋转缓慢蒸发除去溶剂,此过程重复4次,最终得到以APS质量计算涂敷量约为20%(W/W)的手性固定相。
取所得固定相3 g,以正己烷-异丙醇(90∶10,V/V)为匀浆液和顶替液,于约32 MPa压力下装入不锈钢管(150 mm×4.6 mm)中。
2.3 手性固定相的表征
为考察大粒径手性固定相合成时每步反应的键合情况,采用元素分析、红外光谱对其相关参数进行考察。
2.3.1 APS的元素分析。对硅胶及APS进行元素分析,结果为硅胶中氮元素(N)的质量分数W(N)为0%,APS中W(N)为1.25%。从数据看出硅胶上无氮元素,而APS的元素分析结果中含有氮元素,说明3-氨丙基三乙氧基硅烷已键合到硅羟基上。
2.3.2 CDMPC的元素分析和红外光谱分析。纤维素被酯化的程度对CDMPC手性固定相的手性识别能力有直接影响,酯化反应越完全,固定相的手性识别能力越高。
CDMPC的碳(C)、氢(H)、氮元素分析结果[实测值(理论值)]为:W(C)[66.01%(65.66%)],W(H)[6.42%(6.18%)],W(N)[6.67%(6.98%)],实测值与理论值吻合较好,说明纤维素的酯化较完全。
再比较纤维素和CDMPC的红外光谱图,详见图4。
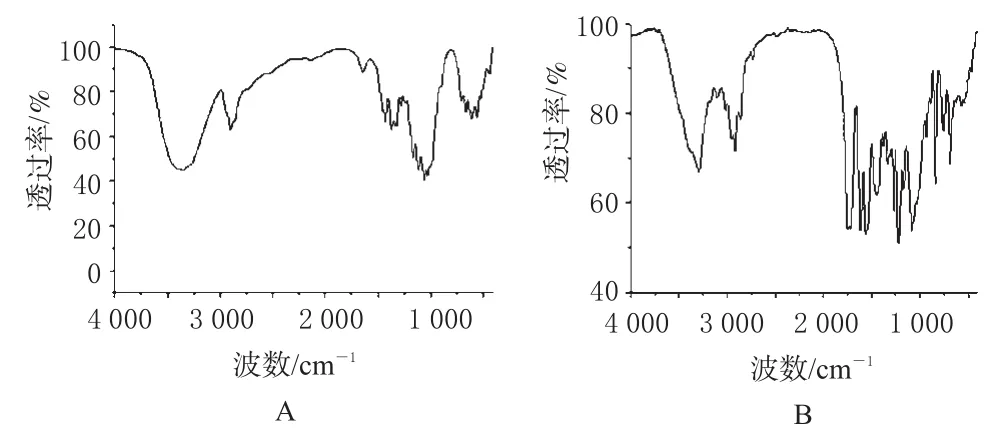
图4 红外光谱图Fig 4 IR spectrum
由图4可见,纤维素在3 346.1 cm-1处—OH的吸收很强,而CDMPC在此处—OH的吸收峰已经很弱,说明纤维素上的羟基已基本被酯化;同时在图4B中,1 724.3 cm-1出现苯基氨基甲酰基的—CO吸收峰,1 615.2、1 559.7 cm-1是苯环的骨架结构,由此说明纤维素上的—OH已基本被3,5-二甲基苯基异氰酸酯酯化。
2.3.3 CDMPC手性固定相的元素分析。按“2.2.3”项下涂敷的质量分数为20%的CDMPC手性固定相的元素分析结果为:W(C)15.55%,W(H)2.13%,W(N)2.14%。而通过理论计算当涂敷量为20%时,CDMPC手性固定相中W(N)的理论值应为2.15%,与实测值吻合较好,说明CDMPC已按20%的涂敷量涂敷于APS上。
2.4 手性拆分性能评价
以普萘洛尔为对象,以合成的CDMPC手性固定相为手性柱,通过对色谱条件的优化,评价在最优的色谱条件下,手性柱的手性拆分性能。
2.4.1 色谱条件。色谱条件:色谱柱:自制的大粒径CDMPC手性色谱柱(150 mm×4.6 mm,40~60µm);流动相:正己烷-异丙醇-三乙胺(不同比例);紫外检测波长:276 nm;流速:0.3、0.5、0.7、0.9 mL·min-1;进样量:20 μL;分离均在室温下进行;色谱柱的死时间(t0)用1,3,5-三叔丁基苯测定。
2.4.2 流动相比例的优化。在涂敷型纤维素衍生物类手性固定相中,通常采用正己烷、异丙醇的正相流动相系统,而普萘洛尔为碱性药物,在流动相中添加适当的碱性溶液有利于其分离和改善峰形。因此,本试验考察了碱性添加剂三乙胺浓度为0.1%时,流动相中正己烷、异丙醇的不同比例对普萘洛尔手性拆分的影响,以2个对映体的保留时间tR1和tR2及根据公式k1'=(tR1-t0)/t0、k2'=(tR2-t0)/t0而得的2 个对映体的容量因子k'、分离因子α和分离度Rs为指标确定最优的流动相组成比例,考察结果详见表1。
由表1显示,随着流动相中异丙醇比例的减少,容量因子k'均变大,表明样品在手性固定相中的保留时间延长;同时分离度增加,表明异丙醇质量比例的减少有利于样品得到更佳的分离效果;分离因子α随之增加,表明固定相对样品对映体的分离效果不断增强。
综合考虑分析时间和分离效果,确定正己烷-异丙醇-三乙胺(95∶5∶0.1,V/V/V)为最佳流动相组成比例。
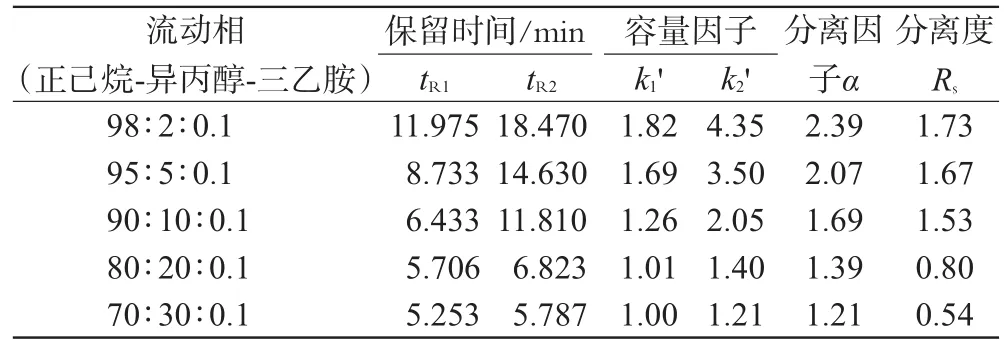
表1 流动相中正己烷、异丙醇的不同比例对分离的影响Tab 1 Effect of different proportions of n-hexane and isopropanol in mobile phase on separation
2.4.3 流速的优化。考察了流动相流速分别为0.3、0.5、0.7、0.9 mL·min-1时对分离的影响,结果,流速与分离度间的关系见图5。
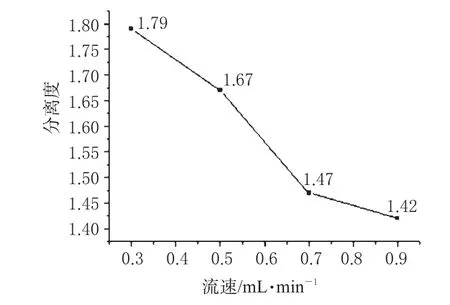
图5 流速对普萘洛尔分离度的影响Fig 5 Effect of flow rate on separation rate of propranolol
由图5可见,流速从0.9 mL·min-1减小到0.5 mL·min-1时,分离度增大,达到基线分离。这是由于对映体在纤维素衍生物上的手性识别作用能量差极小,对映体间的完全分离有赖于溶质与固定相之间的多次相互作用。流速从0.5 mL·min-1减小到0.3 mL·min-1时,虽然分离度有所增加,但色谱峰加宽,分析时间延长,柱压上升。综合考虑分离效果、分析时间和柱压等各方面因素,选择0.5 mL·min-1为较适宜流速。
通过对流动相、流速的优化,最后确定了普奈洛尔分离的流动相为正己烷-异丙醇-三乙胺=95∶5∶0.1(V/V/V,使用前均经0.22 µm 滤膜过滤并超声脱气15 min),流速为0.5 mL·min-1。2.4.4 样品拆分。此条件下样品的手性拆分色谱见图6(其中,2个对映体的确定是通过旋光仪测定区分)。
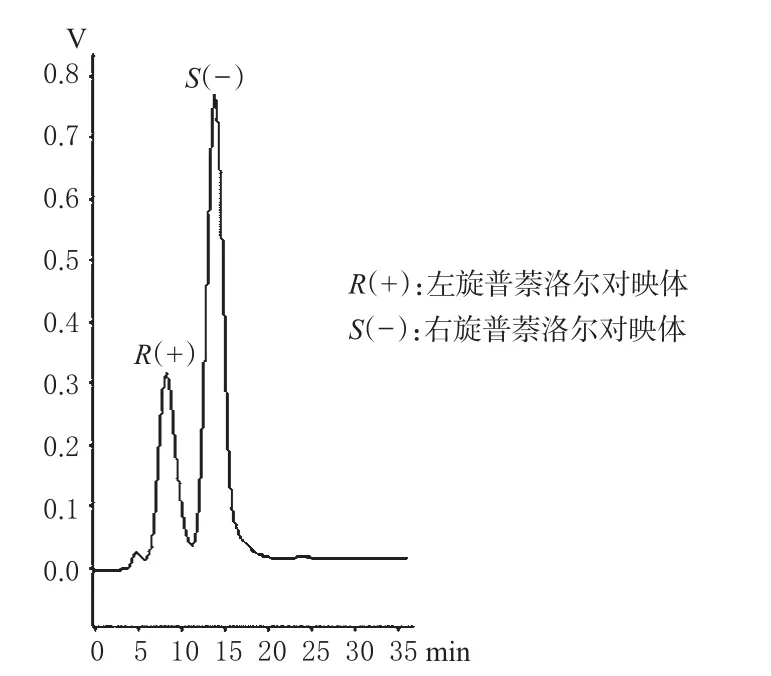
图6 普萘洛尔的手性拆分图谱Fig 6 Chromatogram of chiral separation of propranolol
2.5 手性柱的稳定性
将所制备的CDMPC手性柱在正相条件下连续使用2个月(约450 h),共进样约900针,考察柱压和分离度的变化。结果,柱压由386 psi变为435 psi,变化率为12.7%,表明柱压值一直较低且无明显升高;分离度由1.67变为1.62,分离度下降了2.99%,表明手性拆分能力及柱效均无明显下降现象,这说明制备的CDMPC手性柱具有良好的稳定性。
2.6 CDMPC拆分普萘洛尔的机制分析
由于CDMPC手性固定相是具有左旋三重(3/2)螺旋型空穴的光学活性分子,3,5-二甲基苯基氨基甲酸酯基团围绕纤维素主链形成许多手性空腔,在手性空腔中,靠近纤维素主链的外侧是芳基,内侧是手性固定相的氨基甲酸酯残基,对手性化合物有很好的选择性。此外,纤维素类固定相的螺旋型空穴对进入空腔的芳环部分有很高的亲和力,形成“立体配合”的包结作用[12]。普萘洛尔手性中心附近含有羟基和氨基,当其调整构象进入螺旋槽中,其结构中的羟基和氨基可与CDMPC酰胺键产生氢键作用,萘环与CDMPC中的苯环存在π-π作用,甲基与CDMPC中的甲基形成范德华力,从而与固定相形成缔合物,由于2个对映异构体所形成的缔合物的稳定性不同而达到手性分离。普萘洛尔在CDMPC手性凹槽内的超分子键联示意图见图7。
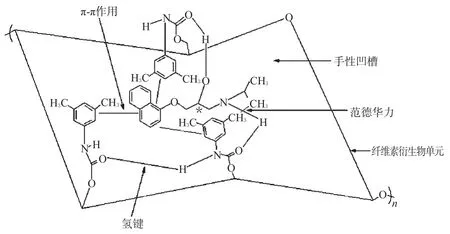
图7 普萘洛尔在CDMPC手性凹槽内的超分子键联示意图Fig 7 Diagram of supra-molecular bindings of propranolol in chiral groove of CDMPC
由图7显示,普萘洛尔能与CDMPC形成3个π-π作用、2个氢键作用和1个范德华力,从而使普萘洛尔在此固定相上获得了较好的手性拆分[13]。
3 讨论
纤维素衍生物类手性固定相由于其拆分的手性化合物范围广,样品载样量大,非常适合于手性药物的制备分离。但由于常规的小粒径(5µm)手性柱存在柱压高、易堵塞、寿命较短等缺点,限制了其在制备分离上的应用。因此,本文采用大粒径硅胶颗粒(40~60µm)制备了涂敷型CDMPC约45 g,达到半制备色谱柱所需的填料要求。将3 g该填料装填在不锈钢管中获得涂敷型CDMPC手性分析柱,并通过对普萘洛尔对映体的分离评价了其手性拆分性能。结果在正相条件下普萘洛尔对映体的分离度达1.67,表明合成的CDMPC填料具有良好的手性拆分性能。
通过以大粒径硅胶颗粒为填料基质,不仅降低了小粒径手性柱制备中硅胶基质的原料费用,同时,由于填料孔径大、填料间的空隙大,减小了对流动相的阻力,因此有效地避免了小粒径手性柱柱压高、易堵塞等缺点,在制备纤维素类手性柱的应用上显示出了更大的优势,在手性药物的制备分离上显示了很好的应用前景。
[1] 周志强,邱 静,江树人,等.涂敷纤维素类手性固定相对甲霜灵中间体的高效液相色谱拆分[J].分析测试学报,2003,22(1):89.
[2] Yashima E.Polysaccharide-based chiral stationary phases for high-performance liquid chromatographic enantioseparation[J].J Chromatogr A,2001,906(1/2):105.
[3] Török G,Goetelen L,Luyckx R.Evaluation of the performance of commercially available polysaccharide-based chiral stationary phases after multicycle operation in multimodal elution mode[J].J Pharmaceut Biomed,2005,39(3/4):425.
[4] Kubota T,Yamamoto C,Okamoto Y.Chromatographic enantioseparation by cycloalkylcarbamate derivatives of cellulose and amylose[J].Chirality,2002,14(5):372.
[5] Kubota T,Yamamoto C,Okamoto Y.Reversed-phase liquid chromatographic enantioseparation by cycloalkylcarboxylates of cellulose and amylose[J].Chirality,2004,16(5):309.
[6] 何永祝,庞 浩,廖 兵.纤维素衍生物手性固定相研究进展[J].化学进展,2006,18(7/8):957.
[7] 韩小茜,温晓光,管永红,等.键合纤维素(4-甲基苯甲酸酯)手性固定相的制备及手性拆分[J].分析化学,2004,32(10):1 287.
[8] Okamoto Y,Kaida Y.Resolution by high-performance liquid chromatography using polysaccharide carbamates and benzoates as chiral stationary phases[J].J Chromatogr A,1994,666(1/2):403.
[9] 陈立仁.液相色谱手性分离[M].北京:科学出版社,2006:72.
[10] 陈小明,杨 利,邹汉法,等.键合型纤维素-苯基氨基甲酸酯手性固定相的制备及用于对映异构体拆分[J].分析化学,2000,28(9):1 074.
[11] 侯经国,周志强,陈立仁,等.纤维素-三(苯基氨基甲酸酯)涂敷手性固定相的制备及其在反相条件下的手性分离[J].色谱,1998,16(4):337.
[12] 许志刚,艾 萍,周 岩,等.纤维素三(3,5-二甲基苯基氨基甲酸酯)液相色谱固定相的制备及手性拆分应用
[J].化学世界,2005,46(12):722.
[13] 刘月启,刘 霞,蒋生祥,等.联苯类对映体在纤维素类手性固定相上的高效液相色谱法直接拆分[J].化学学报,2000,58(11):1 424.
猜你喜欢
井冈山大学学报(自然科学版)(2021年3期)2021-09-10
导航与控制(2021年3期)2021-09-04
天津医科大学学报(2021年4期)2021-08-21
石油化工(2021年3期)2021-04-08
江苏船舶(2019年1期)2019-05-15
沈阳化工大学学报(2018年1期)2018-05-30
中成药(2017年9期)2017-12-19
印刷技术·数字印艺(2015年10期)2015-12-10
医药导报(2015年6期)2015-02-10
中国信息化·学术版(2013年7期)2013-09-03
