
非甾体类5α-还原酶抑制剂研发近况
2011-08-06 00:56廖清江
药学进展 2011年10期
孙 洁, 赵 维, 向 华, 廖清江
(中国药科大学药物化学教研室,江苏 南京,210009)
良性前列腺增生(BPH)是指前列腺间质和上皮细胞的良性腺瘤性增生,为中老年男性常见的一种慢性疾病。其所致前列腺腺体的进行性肿大使尿道狭窄,引起膀胱尿液流出受阻,临床表现为尿急、尿频和尿排不尽,常导致尿道感染、尿潴留、膀胱感染、膀胱结石和肾功能衰竭[1]。BPH的确切病因及发病机制至今仍未完全阐明,目前较为公认的是其为体内雄激素水平异常所致。大量研究显示,雄激素在体内保持正常循环水平,是保证前列腺健康生长及各项功能的必要条件。睾酮是由睾丸及肾上腺产生的一种主要雄激素,在体内可被5α-还原酶和烟酰胺腺嘌呤二核苷酸(NADPH)还原代谢为活性更高的二氢睾酮(DHT),二者均为维持男性显著特征和促进性成熟的重要雄激素[2]。虽然男性血浆内睾酮水平随着年龄的增长而下降,但BPH的患病率却随着年龄的增长而上升,其原因在于老年人前列腺内雄激素总水平仍然很高,其中90%为DHT。DHT对人体前列腺的生长和发育起重要作用,但如果体内DHT水平过高,将会导致内分泌失调,引发多种疾病,如BPH、痤疮、男性秃发和女性多毛症等[3]。
BPH的早期治疗方法主要是前列腺切除术,此疗法虽可有效治疗BPH,但治疗费用高,且有研究表明,前列腺参与人体某些生理活动的调节,切除前列腺的患者常会发生性功能障碍、尿失禁等[4]。前列腺除了通过分泌前列腺液参与精液的形成之外,还能产生多种免疫球蛋白,合成具有抗菌作用的含锌多肽,并具有保护生殖系统免遭细菌和其他病原微生物侵袭的局部免疫功能,故在可能的情况下应尽量保留前列腺[5]。因此,药物治疗成为轻、中度BPH及腺体较大不宜手术患者的主要疗法。BPH发病机制研究表明,体内睾酮是在5α-还原酶和NADPH的联合作用下还原为DHT,进而引发BPH。由此,人们将5α-还原酶作为治疗BPH的重要靶点之一,设计并利用 5α-还原酶抑制剂与睾酮和NADPH竞争性或非竞争性作用于5α-还原酶及其与NADPH的复合物,抑制DHT的生成,从而降低血浆中DHT水平,减少BPH的发生。目前已开发的5α-还原酶抑制剂包括甾体和非甾体两类,其中甾体类以氮杂结构为主,代表药物为非那雄胺(1)和度他雄胺;此外,还有以爱普列特(2)为代表的3-羧酸甾体类抑制剂。以上这些药物均已上市,虽然它们用于BPH的疗效尚佳,但其副作用,如性功能障碍、男子乳腺发育、肌肉组织生长阻碍等也给患者带来极大困扰。据文献报道,此类副作用主要源自于甾体母核,故而非甾体类5α-还原酶抑制剂的研发引起极大关注[6]。
1 5α-还原酶
甾体5α-还原酶是定位于雄激素靶细胞核膜上的膜蛋白,依赖NADPH作为供氢体,催化一系列甾体底物,如睾酮、17α-羟基黄体酮、雄烯二酮等的还原反应[7]。5α-还原酶又分为Ⅰ型和Ⅱ型同工酶(以下简称Ⅰ型和Ⅱ型酶),二者在人体内的分布不同,功效各异。Ⅰ型酶的最适pH值为6.0~8.5,主要存在于皮脂腺和肝脏中,故其抑制剂常用于治疗斑秃、男性脱发、痤疮及妇女多毛症等内分泌疾病;Ⅱ型酶的最适pH值为5.5,主要存在于前列腺、精囊、附睾、毛囊及肝脏中,其抑制剂常用于治疗前列腺疾病,如 BPH[8-9]。
在催化睾酮还原为活性更强的DHT过程中,5α-还原酶起着关键作用。据文献报道,5α-还原酶(E)的还原机制是,其先与氢供体NADPH结合形成二元复合物(E-NADPH),再与睾酮(T,3)结合形成三元复合物(E-NADPH-T)(其间,由于NADPH与5α-还原酶的活性部位发生强烈作用而结合并形成碳正离子,而睾酮3位的羰基具有亲核性,导致三者结合),随后通过电子转移作用,引发睾酮的5位选择性还原,氢原子由NADPH转移至睾酮5位的α面,从而形成DHT的烯醇式盐,接着此烯醇式盐捕获一个质子,脱离NADP+与5α-还原酶的二元复合物(E-NADP+),形成 DHT(4),最后 E-NADP+再分解释放出NADP+与5α-还原酶,进而参与下一次还原作用[10-11](见图1)。


图1 5α-还原酶还原睾酮的机制Figure 1 Mechanism of reduction of testosterone by 5α-reductase
2 非甾体类5α-还原酶抑制剂
非甾体类5α-还原酶抑制剂的设计思路大多源自于模拟甾体结构,即去除甾体结构中的一个或多个环,并加以进一步的结构修饰。
2.1 模拟甾体类
2.1.1 模拟氮杂类 氮杂类是甾体类5α-还原酶抑制剂的主要结构类型之一,对于该类结构的改造方式主要是去除甾体的D环结构,并对C环进行芳构化。Lilly公司首次模拟4-氮杂甾体5α-还原酶抑制剂,研发出苯并喹诺酮类衍生物,该类化合物大多为Ⅰ型酶抑制剂,其中化合物5显示出良好的Ⅰ型酶抑制活性,IC50为17 nmol·L-1。在对化合物5作进一步结构修饰时,以硫原子替换3位上氧原子,则化合物活性明显下降,如化合物 6 的 IC50为 183 nmol·L-1,表明内酰胺结构更利于化合物与5α-还原酶结合;而在B环引入双键也会降低化合物活性,如化合物7和8的IC50分别为120和377 nmol·L-1;当10位甲基被氢替换,会导致化合物活性大幅下降,如化合物9的IC50为 560 nmol·L-1。由此可见,内酰胺结构、10位甲基及化合物的饱和度对该类化合物的Ⅰ型酶抑制活性起关键作用。此外,以反式苯乙烯基替换化合物5的苯环上氯原子所得化合物10,除了具Ⅰ型酶抑制活性(IC50=23 nmol·L-1)之外,对Ⅱ型酶也表现出一定的抑制活性(IC50=180 nmol·L-1),为双重 5α-还原酶抑制剂[12]。
Hosoda等[13]在先导物 4-MA(11)的结构基础上,以3,3-二苯基戊烷为桥梁连接甾体A环与17β侧链,并将甾体A环打开,所得化合物12对Ⅰ型酶具有微弱的抑制活性,在 10 μmol·L-1浓度下的抑制率为22%;当对化合物12作进一步修饰时,将2和2'位的甲基替换为氢原子,所得化合物13在相同浓度下对Ⅰ型酶的抑制率上升至57%,IC50为9.2 μmol·L-1,表明对苯环进行甲基化会降低化合物活性;再对化合物13进行修饰时,将1'位则链上两乙基用丙基替换,所得化合物14和15的活性明显提高,IC50分别为 2.9 和 4.0 μmol·L-1,而当乙基被苄基替换,所得化合物16的活性提高约10倍,IC50为 0.84 μmol·L-1,由此可见,1'位侧链上 R3为较大的疏水性基团时,有利于化合物活性的提高。

10-氮杂甾体类也是氮杂类5α-还原酶抑制剂的另一主要结构类型,其相应的非甾体模拟物亦具有5α-还原酶抑制活性。此类抑制剂有两种基本母核:一种为4a-H系列,即结构中含有1,2-双键;另一种为1-H系列,即结构中含有4,4a-双键(见图3)。
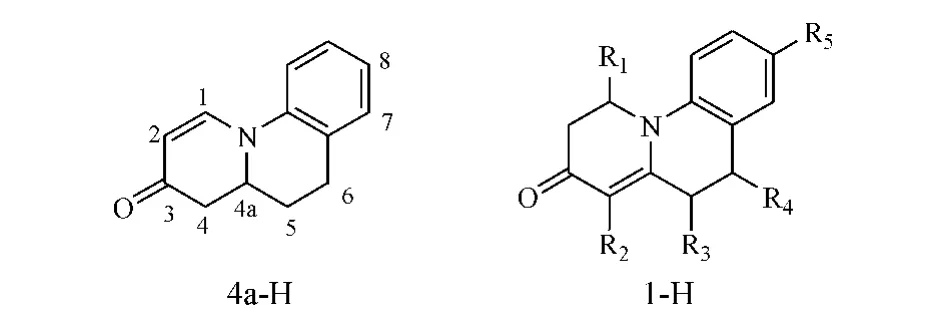
图3 模拟10-氮杂甾体类的非甾体类5α-还原酶抑制剂4a-H系列和1-H系列母核Figure 3 4a-H and 1-H mother nucleus of nonsteroidal 5α-reductase inhibitors analogous to 10-azasteroids
此类化合物大多为Ⅰ型酶抑制剂,其构效关系研究显示,1-H系列化合物的活性优于4a-H系列,其母核1位上不宜有取代基,若1位被甲基等烷基侧链取代,会导致化合物活性明显下降;母核4位与6位引入甲基有利于化合物活性的提高;母核5位不宜引入烷基侧链,否则会带来极大的空间位阻,阻碍化合物与酶的结合,从而显著降低化合物活性;母核8位取代基为甲基或氯、溴等卤素原子,有利于化合物活性提高。例如,化合物17~19的IC50分别为14.4、7.6 和 8.5 nmol·L-1,均为良好的Ⅰ型酶抑制剂[14-15]。

Occhiato研究小组先后研制出母核8位为各种不同取代侧链的1-H系列化合物,结果发现,取代侧链为醚键类或杂环类时,均会削弱化合物对Ⅰ型酶的抑制活性,而苯乙炔基或苯乙烯基侧链则有利于化合物活性的提高,如化合物20和21,对Ⅰ型酶的IC50分别为8.75 和14.2 nmol·L-1;侧链为羧酸苯酯类时,化合物对Ⅰ型和Ⅱ型酶具有良好的双重抑制活性,如化合物22~24对Ⅰ型酶的IC50分别为93、160 和 138 nmol·L-1,对Ⅱ型酶的 IC50分别为 119、134 和 166 nmol·L-1[16-17]。

Hu等[18]报道了具有 5α-还原酶抑制活性的一系列喹诺酮类化合物,其结构类似于甾体的A、B环,并在母核6位引入苯基,以模拟甾体D环结构。其中,化合物25具有良好的Ⅱ型酶抑制活性,在10 μmol·L-1浓度下的抑制率达83%,而对Ⅰ型酶的抑制率只有27%;但若在其1位氮原子处引入甲基,如化合物26,则在相同浓度下对于Ⅱ型酶抑制率降至9%,却对Ⅰ型酶的抑制活性大幅提高,抑制率达87%;此外,苯环上酰胺侧链的结构亦会极大影响化化合物活性,若以异丁基和环己基替代化合物25中苯甲酰胺侧链上的的异丙基,所得化合物27和28对Ⅰ型和Ⅱ型酶均具有良好的抑制活性,在浓度为 10 μmol·L-1时,对Ⅰ型酶抑制率分别为 73% 和82%,对Ⅱ型酶抑制率分别为70%和63%。

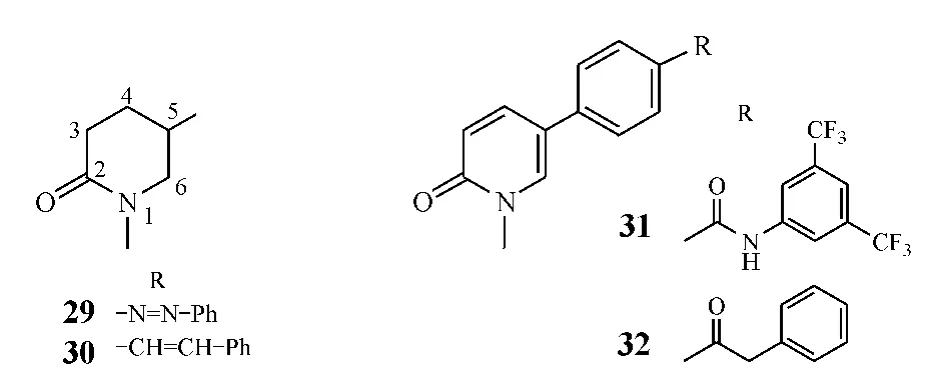
目前研制的模拟氮杂类5α-还原酶抑制剂大多为三环结构,也有类似于甾体A、C环的二环结构,如哌啶酮类、吡啶酮类、联苯类等,但其活性均不如三环结构化合物。其中,哌啶酮类结构特点为2-哌啶酮母核在5位与苯环连接,如化合物29和30的活性较好,对Ⅰ型酶的 IC50分别为 302 和 107 nmol·L-1,对Ⅱ型酶的 IC50分别为 579 和 617 nmol·L-1[12];吡啶酮类结构特点为2-吡啶酮母核在5位与苯环连接,如化合物 31 对Ⅰ型酶的 IC50为 2.6 μmol·L-1[19],化合物32在10 μmol·L-1浓度下对Ⅰ型酶的抑制率为61%[20],均为良好的Ⅰ型酶抑制剂。
2.1.2 模拟爱普列特类 爱普列特是一种新型非竞争性5α-还原酶抑制剂,可选择性抑制Ⅱ型酶,对Ⅰ型酶作用较弱,因此仅作用于前列腺,使其DHT水平下降,而对血液循环中睾酮和DHT水平无甚影响。大多数三环羧酸类非甾体5α-还原酶抑制剂的设计均是模拟爱普列特结构,并去除甾体B、D环。
联苯羧酸结构类似于爱普列特的A、C环结构,其经侧链结构修饰,所得一系列化合物亦具有Ⅱ型酶抑制活性,如化合物33~36的IC50分别为10、4.0、0.64 和 0.22 μmol·L-1。构效关系研究显示,该类化合物的活性随苯环侧链取代基上R基团空间体积的增大而提高[19〛。

2.2 苯甲酸类抑制剂
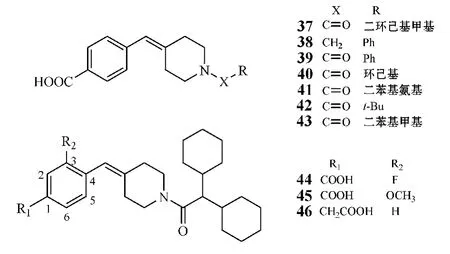
Hartmann 课题组报道了一类具有良好5α-还原酶抑制活性的含苯甲酸结构的非甾体化合物,其中化合物37对于人体Ⅱ型酶抑制活性(IC50=0.06 μmol·L-1)近似于非那雄胺(IC50=5 nmol·L-1),且在10 μmol·L-1浓度下对人体Ⅰ型酶抑制率达46%。此类抑制剂的构效关系研究显示,哌啶环酰胺侧链为化合物活性必需基团,如化合物38中酰胺羰基被还原为亚甲基后,其活性即消失;此外,酰胺侧链的刚性与所占空间大小直接影响化合物的活性,如化合物39~43对人体Ⅱ型酶的IC50分别为9.3、1.65、0.83、3.1 和 0 .53 μmol·L-1,均不如化合物37,尤以化合物39、40、42活性更差,由此可见,酰胺侧链空间结构越大越利于化合物活性的提高,但其刚性不宜过强。故而,该课题组为进一步改善此类抑制剂活性,以二环己酮甲基为酰胺侧链,对苯甲酸基团进行一系列修饰,如于苯环3位引入氟原子或甲氧基。药理实验证实,氟原子的引入可使该类化合物对Ⅱ型酶的抑制活性提高,而对Ⅰ型酶的抑制活性降低,如化合物44对Ⅱ型酶的 IC50为0.011 μmol·L-1,而其即使在 10 μmol·L-1浓度下对Ⅰ型酶的抑制率也只有12%;甲氧基的引入则会同时降低化合物对Ⅰ型与Ⅱ型酶的抑制活性,如化合物 45 对 Ⅱ 型酶的 IC50为 0.13 μmol·L-1,而在10 μmol·L-1浓度下对Ⅰ型酶的抑制率仅为 5%;若将苯甲酸改为苯乙酸基团,所得化合物46对于Ⅱ型酶的抑制活性提高了10 倍(IC50=0.006 μmol·L-1),为良好的Ⅱ型酶抑制剂,但在 10 μmol·L-1浓度下对Ⅰ型酶的抑制率只有 6%[6,21]。
另一类具有Ⅱ型酶抑制活性的苯甲酸系列化合物是以二苯甲酮为母核。其构效关系研究显示:苯甲酸基团中羧酸碳链的长度会影响化合物活性,其中苯乙酸基团替换苯甲酸基团后,化合物活性提高,但碳链过长反而会导致化合物活性下降,如化合物47~49 的 IC50分别为 0.053、0.023 和 2.8 μmol·L-1,而于苯乙酸2位引入氟原子,对化合物活性的影响不大,如化合物50的活性与化合物48相近;二苯甲酮母核对化合物活性的产生极为重要,其中羰基若被还原为亚甲基,化合物活性明显下降,如化合物51在10 μmol·L-1浓度下对Ⅱ型酶的抑制率仅为12%,而对侧链苯氧基进行结构修饰,将4″位取代基变为溴原子,所得化合物52活性明显提高,与非那雄胺的活性相同(IC50=0.005 μmol·L-1)[22]。

2.3 天然产物衍生物
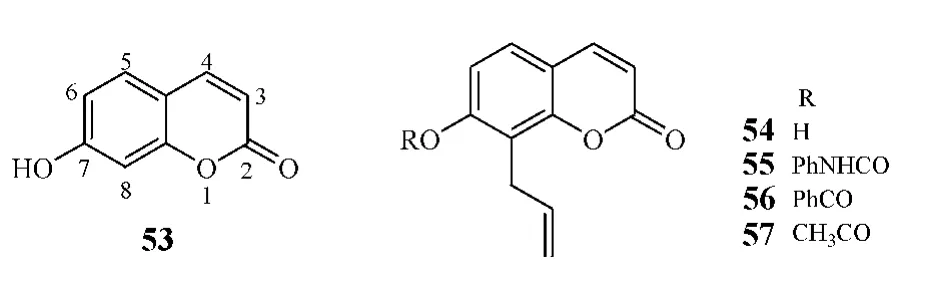
近年来,多种天然产物衍生物被发现具有5α-还原酶抑制活性,故以天然产物为母核进行结构修饰,已成为研发新型非甾体5α-还原酶抑制剂的新思路。Fan课题组以香豆素衍生物伞形酮(7-羟基香豆素,53)为母核,进行结构修饰,研制出一系列Ⅰ型酶抑制剂,其中化合物54~57的IC50分别为0.99、0.62、0.49 和 0.82 μmol·L-1。构效关系研究显示,苯环8位烯丙基侧链为该系列化合物的活性必需基团,且7位取代基上R基团中含有酯键结构有利于化合物活性的提高[23]。
表没食子儿茶素没食子酸酯(58)是绿茶中一种含量丰富的儿茶素,具有抗氧化、抗炎症、抗菌、抗病毒和抗癌作用。近期有研究报道,此类天然物质亦具有抑制5α-还原酶活性,而为提高其活性,以其为母核进行结构修饰,如用多种饱和或不饱和脂肪酸替代母核1'位邻苯三酚侧链,结果显示,在不同饱和脂肪酸侧链中以棕榈酸侧链取代的化合物59活性最佳,其 IC50为 0.53 μmol·L-1;在各种不饱和脂肪酸侧链中,以棕榈油酸侧链取代的化合物60活性最佳,其 IC50为 0.48 μmol·L-1[24]。

3 结语
非甾体5α-还原酶抑制剂因其结构多样性和无激素活性等特点而被广泛开发用于BPH治疗,很多设计构思巧妙、结构新颖和活性较高的该类抑制剂被相继报道,为5α-还原酶抑制剂的研发提供了新的策略和启示。笔者所在实验室亦合成了一系列结构新颖的5α-还原酶抑制剂,并测定了其体外5α-还原酶抑制活性及雄激素受体抑制活性,对其的进一步研究仍在进行中。有理由相信,随着研发工作的不断深入,定会设计出更多的选择性更高、不良反应更小的新型5α-还原酶抑制剂。
[1]Montorsi F,Alcaraz A,Desgrandchamps F,et al.A broader role for 5ARIs in prostate disease?Existing evidence and emerging benefits[J].The Prostate,2009,69(8):895-907.
[2]Mohler J L,Titus M A,Bai S,et al.Activation of the androgen receptor by intratumoral bioconversion of androstanediol to dihydrotestosterone in prostate cancer[J].Cancer Res,2011,71(4):1486-1496.
[3]陈业刚.代谢综合征与良性前列腺增生[J].中华男科学杂志,2010,16(12):1117-1119.
[4]Nickel J C,Méndez-Probst C E,Whelan T F,et al.2010 Update:Guidelines for the management of benign prostatic hyperplasia[J].Can Urol Assoc J,2010,4(5):310-316.
[5]Patel A K,Chapple C R.Medical management of lower urinary tract symptoms in men:current treatment and future approaches[J].Nat Clin Pract Urol 2008,5(4):211-219.
[6]Picard F,Baston E,Hartmann R W,et al.Synthesis of N-substituted piperidine-4-(benzylidene-4-carboxylic acids)and evaluation as inhibitors of steroid-5α-reductase type 1 and 2[J].Bioorgan Med Chem,2000,8(6):1479-1487.
[7]Yamaguchi O,Aikawa K,Shishido K,et al.Current status of 5α-reductase inhibitors in the management of lower urinary tract symptoms and BPH[J].World J Urol,2009,27(6):723-728.
[8]Wilborn C,Taylor L,Poole C,et al.Effects of a purported aromatase and 5α-reductase inhibitor on hormone profiles in college-age men[J].Int J Sport Nutr Exe,2010,20(6):457-465.
[9]Cabeza M,Zambrano A,Heuze I,et al.Novel C-6 substituted and unsubstituted pregnane derivatives as 5α-reductase inhibitors and their effect on hamster flank organs diameter size[J].Steroids,2009,74(10/11):793-802.
[10]Aggarwal S,Thareja S,Verma A,et al.An overview on 5α-reductase inhibitors[J].Steroids,2010,75(2):109-153.
[11]Aggarwal S,Thareja S,Bhardwaj T R,et al.3D-QSAR studies on unsaturated 4-azasteroids as human 5α-reductase inhibitors:a self organizing molecular field analysis approach[J].Eur J Med Chem,2010,45(2):476-481.
[12]Abell A D,Prince M J,McNulty A M,et al.Simple bi-and tricyclic inhibitors of human steroid 5α-reductase[J].Bioorg Med Chem Lett,2000,10(17):1909-1911.
[13]Hosoda S,Hashimoto Y.3,3-Diphenylpentane skeleton as a steroid skeleton substitute:Novel inhibitors of human 5α-reductase 1[J].Bioorg Med Chem Lett,2007,17(19):5414-5418.
[14]Guarna A,Occhiato E G,Scarpi D,et al.Synthesis of 8-chloro-benzo[c]quinolizin-3-ones as potent and selective inhibitors of human steroid 5α-reductase[J].Bioorg Med Chem Lett,2000,10(4):353-356.
[15]Guarna A,Machetti F,Occhiato E G,et al.Benzo[c]quinolizin-3-ones:A novel class of potent and selective nonsteroidal inhibitors of human steroid 5α-reductase 1[J].J Med Chem,2000,43(20):3718-3735.
[16]Occhiato E G,Ferrali A,Menchi G,et al.Synthesis,biological activity,and three-dimensional quantitative structure-activity relationship model for a series of benzo[c]quinolizin-3-ones,nonsteroidal inhibitors of human steroid 5α-reductase 1[J].J Med Chem,2004,47(14):3546-3560.
[17]Ferrali A,Menchi G,Occhiato E G,et al.Synthesis and activity of 8-substituted benzo[c]quinolizin-3-ones as dual inhibitors of human 5α-reductases 1 and 2[J].Bioorg Med Chem Lett,2005,15(1):145-148.
[18]Hu Q Z,Yin L,Jagusch C,et al.Isopropylidene substitution increases activity and selectivity of biphenylmethylene 4-pyridine type CYP17 inhibitors[J].J Med Chem,2010,53(13):5049-5053.
[19]Picard F,Schulz T,Hartmann R W.5-Phenyl substituted 1-methyl-2-pyridones and 40-substituted biphenyl-4-carboxylic acids.Synthesis and evaluation as inhibitors of steroid-5α-reductase type 1 and 2[J].Bioorgan Med Chem,2002,10(2):437-448.
[20]McCarthy A R,Hartmann R W,Abell A D.Evaluation of 40-substituted bicyclic pyridones as non-steroidal inhibitors of steroid 5α-reductase[J].Bioorg Med Chem Lett,2007,17(13):3603-3607.
[21]Picard F,Barassin S,Hartmann R W,et al.Synthesis and evaluation of 2'-substituted 4-(4'-carboxy-or 4'-carboxymethylbenzylidene)-N-acylpiperidines:highly potent and in vivo active steroid 5α-reductase type 2 inhibitors[J].J Med Chem,2002,45(16):3406-3417.
[22]Salem O I A,Frotscher M,Scherer C,et al.Novel 5αreductase inhibitors:synthesis,structure-activity studies,and pharmacokinetic profile ofphenoxybenzoylphenyl acetic acids[J].J Med Chem,2006,49(2):748-759.
[23]Fan G J,Mar W,Park M K,et al.A novel class of inhibitors for steroid 5α-reductase:Synthesis and evaluation of umbelliferone derivatives[J].Bioorg Med Chem Lett,2001,11(17):2361-2363.
[24]Lin S F,Lin Y H,Lin M,et al.Synthesis and structureactivity relationship of 3-O-acylated(-)-epigallocatechins as 5α-reductase inhibitors[J].Eur J Med Chem,2010,45(12):6068-6076.
猜你喜欢
现代食品(2022年18期)2022-10-12
现代临床医学(2022年3期)2022-06-06
健康体检与管理(2022年4期)2022-05-13
现代农村科技(2022年1期)2022-01-21
天津医科大学学报(2021年4期)2021-08-21
昆明医科大学学报(2021年5期)2021-07-22
食品界(2021年7期)2021-07-19
现代临床医学(2021年2期)2021-03-29
杂草学报(2016年4期)2017-05-31
吉林农业·下半月(2016年11期)2017-01-09
