
噁唑烷酮类抗菌药的研究进展
2011-08-06 05:13尤启冬
药学与临床研究 2011年4期
杨 娜,尤启冬
中国药科大学药物化学教研室,南京 210009
抗生素和合成抗菌药物是目前人类治疗细菌感染性疾病的首选药物。在我国,抗菌药物被过度使用甚至滥用的情况已很突出。据统计,临床上用于预防性抗菌药物处方占抗菌药物总消耗量的50%以上,而其中确实为细菌感染者仅占极少数。抗生素的过多使用甚至滥用,使得细菌耐药问题日益严重。其中,革兰阳性菌(G+)的耐药问题尤为严重,例如耐甲氧西林的金黄色葡萄球菌(MRSA)和表皮葡萄球菌(MRSE),耐青霉素的肺炎链球菌(PRSP)及耐万古霉素的肠球菌(VRE)等,这些耐药菌的出现严重降低了现有药物的疗效,导致患者治疗时间的显著延长和死亡率的提高。在限制抗菌药物临床使用的同时,开发全新结构和独特作用机制的抗菌药物是解决细菌耐药性问题的根本出路[1-2]。
噁唑烷酮类化合物是一类新型的治疗细菌性感染的抗菌药,可抑制蛋白质合成的起始阶段并很少出现交叉耐药性,由于具有独特的作用机制而备受人们关注。第一个噁唑烷酮类抗菌剂—Linezolid(利奈唑胺),已于2000年4月在美国批准上市,用于治疗多重耐药G+菌引起的感染[3]。Linezolid被证明是治疗G+细菌引起的严重感染的一个重要的药物,目前广泛用于治疗由MRSA引起的医院获得性肺炎以及复杂的皮肤和软组织感染。Linezolid的成功上市归功于其不管是静脉注射还是口服都具有高生物利用度和较好的ADME(药物在体内的吸收、分布、代谢及排泄过程)性质。
1 噁唑烷酮类抗菌药的作用机制
噁唑烷酮类抗菌药是一种化学合成的抗菌药物,其对临床上重要的敏感和耐药革兰阳性菌,如MRSA、VRE和PRSP均有抑菌活性。细菌蛋白质合成包括起始、延长及终止阶段,其中,起始阶段需由50S亚基、30S亚基、mRNA及起始型甲酰蛋氨酸tRNA(fMet-tRNA)形成的复合物参与。肽酰转移酶抑制剂(如氯霉素、林可霉素)是通过与核糖体50S亚基结合,抑制肽酰转移酶,进而阻断肽链延长,抑制细菌蛋白质合成。噁唑烷酮类抗菌药的结合位点也位于50S,但其抑制的是起始复合物的形成,该复合物由30S亚基、fMet-tRNA、mRNA、GTP以及起始因子1-3组成,它并不阻碍fMet-tRNA的形成、延长或终止,因此该类药物与其他抗菌药具有不同的作用机制,不会与其他抗菌药发生交叉耐药现象。尽管噁唑烷酮抗菌药并不抑制肽基转移酶,但还是与氯霉素、林可霉素等竞争性的结合50S亚基,该现象表明它们具有相近的结合位点。研究表明,噁唑烷酮类化合物的作用位点与有些蛋白合成抑制剂(如氯霉素、林可霉素类)存在部分重叠。总之,噁唑烷酮类化合物与50S亚基结合的影响就是抑制了70S的形成。如果70S已经形成,与噁唑烷酮类化合物结合就会在肽键形成时抑制肽链从A位到P位的易位[4-5]。
2 噁唑烷酮类化合物的发展

图1 DuP-721
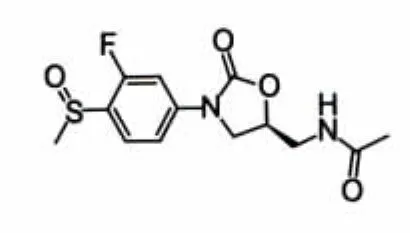
图2 DuP-105
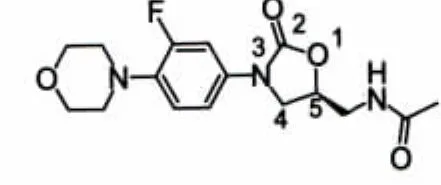
图3 linezolid
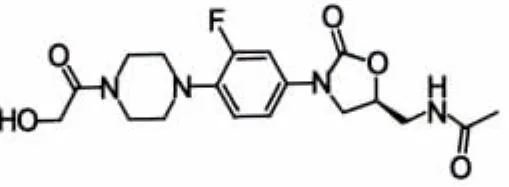
图4 eperezolid
1987年,杜邦(DuPont)公司合成了2个噁唑烷酮类化合物 DuP-721(图 1)和 DuP-105(图 2),临床前研究结果显示,两者对MRSA均具有显著抑菌活性且在大鼠体内药动学性质良好,但后因毒性较大而中止开发。随后,Pharmacia和Upjohn公司通过对该类化合物结构的优化,研制出Linezolid(利奈唑胺,图 3)和 Eperezolid(依哌唑胺,图 4),两者在体内外活性相当,但依哌唑胺在体内清除速率较Linezolid快,需每天服用3次,药动学性质亦不如Linezolid理想,故Linezolid成为目前唯一被美国FDA批准上市的噁唑烷酮类抗菌药物。尽管在使用初期Linezolid显示出令人满意的治疗效果,但长期使用后产生副作用,如单胺氧化酶(MAO)抑制作用,使其与许多药物(如5-羟色胺再摄取抑制剂或其他抗抑郁剂等)无法同时使用,出现了耐药的肠球菌并伴随血小板减少等不良反应,尤其耐药现象日益严重,促使许多药物化学家继续开发具有更高抗菌活性及安全性的噁唑烷酮类抗菌剂以解决这些问题。新研究主要针对扩大该类药物的抗菌谱、重建耐药细菌对该类药物的敏感性以及降低其潜在毒性。表1显示了近年来具有开发潜力的该类化合物及其研究现状。

表1 不同结构的噁唑烷酮类化合物的研究现状
2.1 曾有较大前景,但现已终止研究的化合物
2.1.1 AZD-2563 AZD-2563(posizolid,图 5)是由Astrazeneca公司研制的一系列乙酰胺甲基替换成O或N连接的五元或六元芳香环的噁唑烷酮类衍生物之一[6]。AZD-2563对粪肠球菌、MRSA和甲氧西林敏感金黄色葡萄球菌(MSSA)的MIC值均为1μg·mL-1(Linezolid 对这些菌 MIC 值均为 2μg·mL-1),对MRSE、甲氧西林敏感的表皮葡萄球菌(MSSE)、溶血葡萄球菌、肺炎链球菌的MIC值均为0.5μg·mL-1(Linezolid 均为 1μg·mL-1), 对屎肠球菌的 MIC 值为 2μg·mL-1,与 Linezolid 相当[7]。AZD-2563 具有较好的药代动力学性质:半衰期(t1/2)为 1.05~1.55 h,且抑菌作用具有时间依赖性。与Linezolid不同,该化合物能够产生长期持久的效果。有研究表明AZD-2563对肠球菌和葡萄球菌具有较低的体外突变率[8]。但Astrazeneca公司对其进行Ⅰ期临床研究显示,其各项性质未达到公司预期的效果,因此公司中断了该药的研发。具体的Ⅰ期临床数据未见报道。
2.1.2 Ranbezolid Ranbezolid(RBX-7644,图 6)是由Ranbaxy实验室开发的一类新型广谱噁唑烷酮抗菌药。Ranbezolid是引入了5-硝基呋喃取代亚甲基而得,能特异性地对细菌核糖体产生作用,而对哺乳动物核糖体效果不大,被认为是一个有效地抑制细菌核糖体的药物。在表皮葡萄球菌中研究者观察到Ranbezolid能够抑制细胞壁和脂质的合成。而使用Linezolid的细菌未有该现象,因此,猜测硝基呋喃环会影响细胞膜完整性,这可能是对表葡菌的体外杀菌活性较好的原因之一[9]。该化合物具有良好的水溶性,可口服或静注,且耐受性较好。Ranbezolid对金黄色葡萄球菌、MRSA的MIC值均为1μg·mL-1,活性为Linezolid的两倍;对肠球菌、VRE 的 MIC 值均为 2μg·mL-1,与 Linezolid 相当;对化脓性链球菌、肺炎链球菌的MIC值均为0.125μg·mL-1(Linezolid 均为 2μg·mL-1)[10]。 Ranbezolid 在体内也具有良好的药动学性质和安全性,已于2003年4月完成了Ⅰ期临床研究,但未见其后续报道。但曾有文献报道:含有硝基呋喃基团的其他抗菌化合物可被体内相关酶催化,将硝基转化为活性亚硝基自由基中间体,进而产生细胞毒性。这可能是此类化合物研究终止的原因[1]。
2.1.3 DA-7867 DA-7867(图 7)是由韩国 Dong-A制药公司开发的,其结构中包含了一个联芳环系统,DA-7867对MSSA、万古霉素敏感的粪肠球菌及屎肠球菌、VRE、PRSP 的 MIC 值均为 0.2μg·mL-1(Linezolid 均为 1.56μg·mL-1), 对 MRSA、 耐 Linezolid肠球菌、巴西诺卡氏菌的MIC值分别为0.39、0.5、0.03μg·mL-1,明显优于 Linezolid(MIC 分别为3.13、16、0.5μg·mL-1)[11-12]。 体内试验表明,DA-7867对金黄色葡萄球菌、MRSA、肺炎链球菌、PRSP、VRE的ED50分别为 3.4、2.6、11.6、2.4、4.5 mg·kg-1。 在小鼠模型中,以5 mg·kg-1剂量口服给药DA-7867,其半衰期(t1/2)、生物利用度、最大浓度(Cmax)和曲线下面积(AUC)分别为 5.56 h、91%、4.24μg·mL-1及 32.5μg·h·mL-1。尽管 DA-7867 具有良好的抗菌活性,但因该化合物结构较大,疏水基团较多,导致血清结合率增加,溶解度降低,口服生物利用度降低(与Linezolid相比)。因此,为了解决溶解度和口服活性较低的问题,Dong-A制药公司以该化合物作为先导物,设计了一系列新的化合物,其中Torezolid表现出较好的活性,将其以磷酸盐的前药形式给药,解决了溶解度低及生物利用度较差的问题。Torezolid目前已处于临床Ⅲ期研究中,该化合物将在后文进行介绍。

图5 AZD-2563
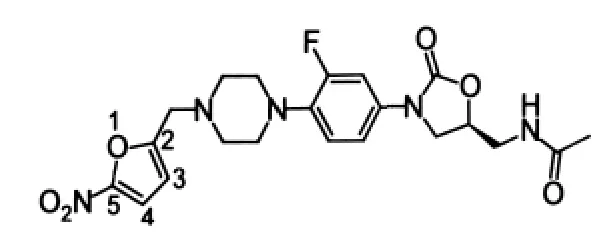
图6 ranbezolid
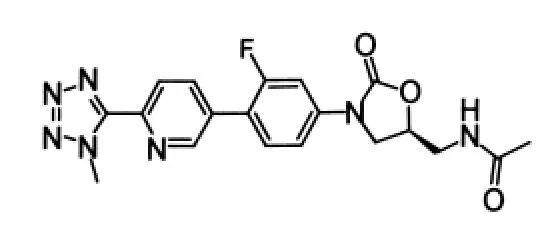
图7 DA-7867
2.2 目前正在临床研究中的化合物
2.2.1 Radezolid Rib-X制药公司的Graham Johnson等[13]分别用Linezolid和非选择性抗菌药稀疏霉素与核糖体50S亚基进行对接,将对接模型所得的活性位点叠加,发现这两个药物在细菌核糖体50S亚基PTC区域的结合位点相近,若能设计出可同时占据这两个相邻结合位点的新化合物,则抗菌活性及抗菌谱都将大大提高,结合计算机分析和化合物结构信息,设计了Rx-01系列化合物。该系列化合物均具有联芳环的结构,对MRSA、MSSA、凝固酶阴性葡萄球菌(CNS)等细菌都有较好的活性,其中 Rx-01-667(RX-1741,Radezolid,图 8)对肺炎链球菌的 MIC低于 0.25μg·mL-1,而对化脓性链球菌、耐Linezolid的粪肠球菌、流感嗜血杆菌、卡他莫拉菌、MSSA 和 MRSA 的 MIC 分别为 0.25、1、0.5、0.5、2 和 4μg·mL-1[14],对下呼吸道感染致病病原菌也有活性,如肺炎支原体(MIC 为 1~2μg·mL-1)、嗜肺军团菌(MIC 为 0.12~0.5μg·mL-1)和肺炎衣原体(MIC 为 1~4μg·mL-1)。因此,该化合物成为第一个上临床的联芳环结构的噁唑烷酮类化合物,用于治疗严重的多药抗药性感染。Radezolid已完成了两个Ⅱ期临床实验:一个是治疗非复杂性皮肤和皮肤结构感染(uSSSI);另一个是社区获得性肺炎(CAP)的治疗[15]。在随机、双盲的Ⅱ期试验中,对160例来自加拿大、俄罗斯和美国的轻至中度社区获得性肺炎病人,给药300和450 mg qd或450 mg bid,连续10天,Radezolid都显示出安全有效的特性,临床治愈率为78%~92%。该化合物对CAP病原体包括肺炎链球菌、MSSA、流感嗜血杆菌和非典型呼吸道病原体显示出较好的活性。它的耐受性良好,最常见的不良反应为轻度的肠胃不适。没有观察到血液学不良反应。
2.2.2 Torezolid Torezolid(TR-700,DA-7157,图 9)及其磷酸酯前药TR-701是由Trius Therapeutics公司研制的新型第二代噁唑烷酮类口服抗菌药。该化合物在DA-7867结构基础上进行结构修饰,并且制成前药TR-701(图10),提高了药物的生物利用度。小鼠大腿感染模型中Torezolid磷酸盐具有杀菌活性。另外,Torezolid对含有cfr(核糖体的甲基转移酶基因)的耐Linezolid的葡萄球菌也有作用。通过Torezolid与DA-7867进行比较发现,将乙酰氨基转换成羟基基本上对活性没有影响或影响较小,而之前,其他羟基化合物观察到活性很弱,可能是由于细菌穿透性/外排等问题。目前,TR-701用于治疗严重复杂性皮肤及皮肤组织感染(cSSSI)的Ⅱ期临床研究已顺利完成[15],并且正在进行对急性细菌性皮肤和皮肤结构感染(ABSSSI)病人进行随机、双盲的Ⅲ期临床实验。在随机、双盲的治疗cSSSI的Ⅱ期临床实验中,给药Torezolid分别以200、300或400 mg qd,持续5~7d,发现这三种剂量都是安全有效的,对这三种剂量临床的治愈率分别为100%、93%和96%,并且在治疗后21-28天的随访中并无临床复发现象,对血小板、肝酶无大影响,该化合物耐受性良好,有轻微不良反应,未见需要停药的不良现象。Torezolid对临床所有的G+菌、某些G-菌以及非典型衣原体属都有效。本品对MRSA和MSSA的MIC值均为 0.5μg·mL-1,抗菌活性优于 Linezolid(MIC 为 4μg·mL-1)[16];对粪肠球菌的MIC值也低于Linezolid(0.5μg·mL-1vs 2μg·mL-1);对耐 Linezolid 的金葡菌的 MIC90为 2μg·mL-1。
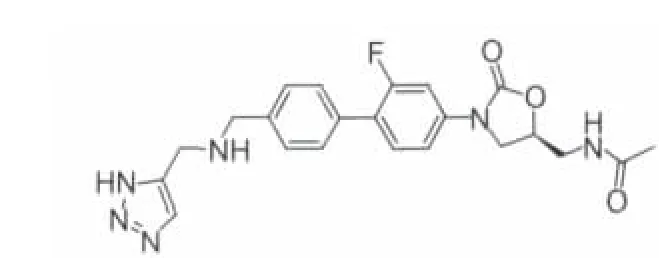
图8 radezolid
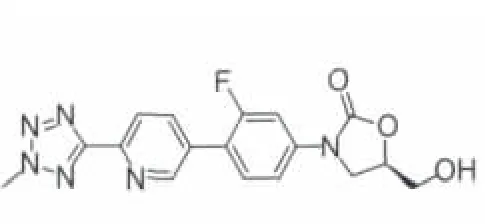
图9 torezolid(TR-700)
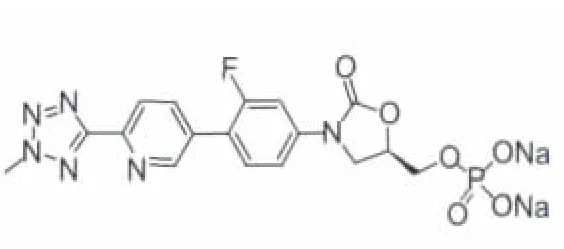
图10 TR-701

图11 PNU-100480
2.2.3 PNU-100480 PNU-100480(图 11)是由辉瑞(之前是Pharmacia)公司开发的一种噁唑烷酮类抗菌药,特别是对结核分枝杆菌具有较好活性,且具有治疗多药耐药和广泛耐药结核病的潜力[17]。该化合物将Linezolid吗啉环上的氧换成了硫,具有更好的抗结核作用。该化合物已完成药物动力学评估和安全单剂量的I期临床试验,并即将进行一个开放式的、随机的、对新诊断的肺结核敏感病人的Ⅱ期临床实验,以评估PNU-100480早期抗菌活性(EBA)和全血活动(WBA)。PNU-100480 对 23 株结核分枝杆菌的平均 MIC 值为 0.12μg·mL-1(Linezolid 为0.39μg·mL-1),活性比 Linezolid 高 3.2 倍,且在体外表现出对多种临床分离的复杂结核杆菌具有很好的疗效(MICs范围为 0.5~4μg·mL-1),对 5 株药物敏感和5株多药耐药结核分枝杆菌的MIC90值均小于 0.5μg·mL-1。
2.3 近两年报道具有优良活性的化合物
2.3.1 RWJ-416457 为了降低Linezolid的耐药性和毒性,Johnson&Johnson Pharmaceutical公司开发了一类新型的吡咯并吡唑基取代的噁唑烷酮类化合物,RWJ-416457(图 12)作为其中代表化合物进行了进一步的研究开发。RWJ-416457的靶点主要是革兰阳性病原体,包括甲氧西林敏感和耐药金黄色葡萄球菌及万古霉素敏感和抗药性的肠球菌等[18]。据报道,该化合物处于I期临床实验阶段[19],但未见任何临床数据报道。体外实验中,它具有较低的突变率[20],对粪肠球菌、屎肠球菌和金黄色葡萄球菌分别为每1000亿中<1.8、<2.9和<1.3的突变株(而 Linezolid 分别为<7.7、<3.2和<1.4/1000 亿)。 该化合物对于全身性葡萄球菌活性是Linezolid的2倍,ED50为 1.5~5 mg·kg-1·d-1。 对于 MSSA 活性较万古霉素弱2~4倍,但对于CA-MRSA (社区获得性MRSA)的皮肤感染活性为万古霉素的4倍,对该两种菌的活性与Linezolid相当。在肺炎球菌模型中,RWJ-416457活性为Linezolid的2~4倍,两者AUC值相当,其t1/2值为Linezolid的3倍[21]。
2.3.2 LCB01-0371 LCB01-0371(该化合物完整结构未见报道,母环结构见图13)是由LegoChem BioSciences公司开发的一个新型的具有环状氨基腙结构的噁唑烷酮化合物,用于细菌性感染的治疗,包括由MRSA引起的感染。体外试验数据表明:该化合物对MSSA、MRSA、卡他莫拉菌、流感嗜血杆菌的活性均为Linezolid的2倍,对CNS、肠球菌、VRE的活性与Linezolid相同[22]。在系统性感染小鼠模型中,口服 LCB01-0371,对 MSSA、MRSA、粪肠球菌、流感嗜血杆菌和肺炎链球菌的ED50值分别为4.71、2.77、4.10、9.96 和 2.28 mg·kg-1。 在 7 天重复的大鼠经口毒性研究中,观察到该化合物的AUC和Cmax具有剂量依赖性,但是没有积聚现象。
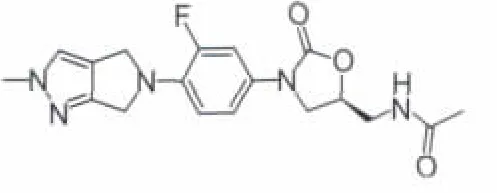
图12 RWJ-416457
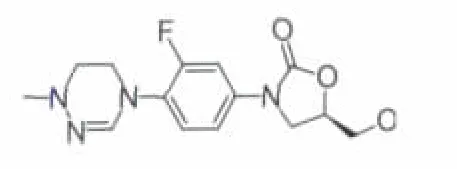
图13 LCB01-0371母环
3 展 望
由于临床上Linezolid耐药菌的出现,以及Linezolid长期使用后产生的副作用,如单胺氧化酶(MAO)抑制作用,并伴随血小板减少和在一些病例中出现的骨髓抑制作用等不良反应,促使药物化学家们继续开发具有更高抗菌活性及安全性的噁唑烷酮类抗菌剂以解决这些问题。每年都有新结构的噁唑烷酮类抗菌药被开发出来,但到目前为止,只有Radezolid和Torezolid仍在进行临床实验,大多数化合物已经停止研究,因此仍然需要科学家对噁唑烷酮类抗菌药物进行更深入的研究。通过对临床化合物性质的研究,特别是对Radezolid和Torezolid结构进行改变,发现联芳结构能够更有效地与细菌核糖体结合,提高化合物的抗菌活性。本课题组结合计算机辅助药物设计,设计并合成了一系列联芳结构的噁唑烷酮类化合物,对抗菌活性有一定的提高。由此可见,联芳类噁唑烷酮化合物具有广大的前景,希望不久的将来会有第二代的噁唑烷酮类抗菌药上市。
[1]杨 燕,尤启冬.噁唑烷酮类抗菌剂构效关系及结构改造研究进展[J].药学进展,2010,34(11):481-90.
[2]周伟澄.作用于细菌蛋白质合成的全合成抗菌剂研究进展[J].中国医药工业杂志,2007,38(11):805-12.
[3]Poulakou G,Giamarellou H.Investigational treatments for postoperative surgical site infections[J].Expert Opin Investig Drugs,2007,16(2):137-55.
[4]Sood R,Bhadauriya T,Rao M,et al.Antimycobacterial activities of oxazolidinones:a review[J].Infect Disord Drug Targets,2006,6(4):343-54.
[5]Howe RA,Woottonet M,Noel AR,et al.Activity of AZD2563,a novel oxazolidinone,against staphylococcus aureus strains with reduced susceptibility to vancomycin or linezolid[J].Antimicrob Agents Chemother,2003,47(11):3651-52.
[6]Johnson AP.AZD-2563 AstraZeneca[J].Curr Opin Investig Drugs,2002,3(6):848-52.
[7]Fluit AC,Schmitz FJ,Verhoef J,et al.In vitro activity of AZD2563,a novel oxazolidinone,against European Gram-positive cocci[J].J Antimicrob Chemother,2002,50(2):271-6.
[8]Stockdale MW,Woodford N,Tysall L,et al.Low in vitro selection frequencies of enterococcal and Staphylococcal mutants resistant to the oxazolidinone AZD2563[J].Int J Antimicrob Agents,2004,23(1):88-91.
[9]Kalia V,Miglani R,Raj VS,et al.Mode of action of Ranbezolid against Staphylococci and structural modeling studies of its interaction with ribosomes[J].Antimicrob Agents Chemother,2009,53(4):1427-33.
[10]Das B,Rudra S,Yadav A,et al.Synthesis and SAR ofnoveloxazolidinones:Discovery ofranbezolid[J].Bioorg Med Chem Lett,2005,15(19):4261-7.
[11]Yoon EJ,Jo YW,Choi SH,et al.In vitro and in vivo activities of DA-7867,a new oxazolidinone,against aerobic gram-positive bacteria[J].Antimicrob Agents Chemother,2005,49(6):2498-500
[12]Vera-Cabrera L,Gonzalez E,Choi SH,et al.In vitro activities of new antimicrobials against nocardia brasiliensis[J].Antimicrob Agents Chemother,2004,48(2):602-4.
[13]Zhou J,Bhattacharjee A,Chen S,et al.Design at the atomic level:Design of biaryloxazolidinones as potent orally active antibiotics[J].Bioorg Med Chem Lett,2008,18(23):6175-8.
[14]Skripkin E,McConnell TS,DeVito J,et al.Rx-01,a new family of oxazolidinones that overcome ribosome-based linezolid resistance[J].Antimicrob Agents Chemother,2008,52(10):3550-7.
[15]Theuretzbacher U.Future antibiotics scenarios:is the tide starting to turn[J].Int J Antimicrob Agents,2009,34(1):15-20.
[16]Livermore DM,Mushtaq S,Warner M,et al.Activity of oxazolidinone TR-700 against linezolid-susceptible and-resistant staphylococci and enterococci[J].J Antimicrob Chemother,2009,63(4):713-5.
[17]Williams KN,Stover CK,Zhu T,et al.Promising antituberculosis activity of the oxazolidinone PNU-100480 relative to that of linezolid in a murine model[J].Antimicrob Agents Chemother,2009,53(4):1314-9.
[18]Foleno BD,Abbanat D,Goldschmidt RM,et al.In vitro antibacterial activity of the pyrrolopyrazolyl-substituted oxazolidinone RWJ-416457[J].Antimicrob A-gents Chemother,2007,51(1):361-5.
[19]Abbanat D,Morrow B,Bush K,et al.New agents in development for the treatment of bacterial infections[J].Curr Opin Pharmacol,2008,8(5):582-92.
[20]Santoro CM,Bush K,Abbanat D.Characterisation of staphylococcus aureus and enterococcus faecalis mutants with reduced susceptibility to the investigational oxazolidinone RWJ-416457[J].Int J Antimicrob A-gents,2010,36(5):424-9.
[21]Hilliard JJ,Fernandez J,Melton J,et al.In vivo activity of the pyrrolopyrazolyl-substituted oxazolidinone RWJ-416457[J].Antimicrob Agents Chemother,2009,53(5):2028-33.
[22]Jeong JW,Kwak JH,Lee HH,et al.In vitro and in vivo activities of LCB01-0371,a new oxazolidinone[J].Antimicrob Agents Chemother,2010,54(12):5359-62.
猜你喜欢
当代化工研究(2022年19期)2022-11-04
纺织标准与质量(2022年2期)2022-07-12
中国典型病例大全(2022年7期)2022-04-22
中国药学药品知识仓库(2022年1期)2022-03-23
中国药学药品知识仓库(2022年2期)2022-03-23
农业与技术(2021年3期)2021-12-06
保健与生活(2019年19期)2019-11-06
科学大观园(2019年23期)2019-09-10
特别健康·下半月(2019年7期)2019-07-29
特别健康·下半月(2017年5期)2017-06-15
