
肌肉萎缩蛋白Fbox-1基因沉默对糖皮质激素作用下肌管萎缩的影响
2011-07-09 07:33王会玲张金元
肾脏病与透析肾移植杂志 2011年1期
王会玲 许 烨 张金元
终末期肾功能衰竭(ESRF)和透析患者常并发进行性蛋白质-能量消耗(protein-energy wasting,PEW),其中骨骼肌消耗(muscle wasting)是导致肾功能衰竭患者虚弱、乏力、生活质量下降的主要原因,也是患者蛋白质-能量性营养不良的主要机制[1]。影响肌肉蛋白质代谢的机制尚不清楚,尿毒症时毒素蓄积、代谢性酸中毒、胰岛素抵抗、微炎症状态等,都可能促进/加速骨骼肌蛋白质分解代谢[2]。文献报道泛素-蛋白酶体系统(ubiquitin proteasome system,UPS)是蛋白质分解的重要执行者,代谢状态下内源性糖皮质激素(glucocorticoids,GC)水平升高,可竞争性拮抗胰岛素受体信号,刺激肌萎缩关键基因表达[3]。我们既往研究显示小鼠慢性肾脏病(CKD)模型的血清GC水平可显著升高10~13倍[4]。本研究通过体外培养肌细胞,研究GC对肌管蛋白质代谢影响及肌肉萎缩蛋白Fbox-1(muscle atrophy F-box,MAFbx,也称为Atrogin-1)的作用机制。
材料和方法
材料与试剂小鼠肌纤维细胞株 C2C12细胞(ATCC A)(晶天生物科技公司);绿色荧光蛋白病毒载体(SMARTvector Empty vector,Dharmacon);DMEM培养液(Cellgrov);胎牛血清(GIBCO),马血清(Sigma);液闪计数器(Beckman);CellLyticTMMT细胞裂解液(Sigma);RNA提取试剂TRIzol Reagent(invitrogen);siRNA-Atrogin-1试剂盒(Dharmacon,ON-Target plus SMARTpool,mouse FBx032,Cat#L-049180-00);Fbx一抗(Everest);地塞米松(dexmethasone,DEX;American Regent Lab Inc);荧光倒置显微镜(Olympus 1x70)。
C2C12体外培养及肌管形态观察C2C12细胞以1×106cell/cm2密度接种于6孔板,用含10%胎牛血清的DMEM,置37℃、5%二氧化碳培养箱中培养,细胞达90%密度时,换含2%的马血清DMEM继续培养,3~4d后细胞融合分化形成肌管,加入GFP 2 μl/孔,24h后荧光倒置显微镜观察肌管已着色,用DEX处理细胞,48h后观察肌管形态并拍照,用ImageJ软件(NIH,Frederick,MD)分析。
3H-酪氨酸同位素标记检测肌管蛋白合成和分解蛋白合成采用3H-酪氨酸(3H-Tyrosine)掺入法检测,在细胞分化形成肌管后第2~3d,每孔加入2.5 μmol/L3H-Tyrosine×30 min,药物处理2h后,DMEM洗3次×10 min,刮下细胞,以10%三氯乙酸(TCA)沉淀,用0.5N纯碱溶解沉淀,进行液闪计数并测定蛋白浓度,结果以cpm/g表示。蛋白分解采用3H-Tyrosine释放率检测,肌管分化好后,加入3H-Tyrosine×20h,用含2%马血清的DMEM洗3遍以除去培养液中的3H-Tyrosine,加入含2 mmol/L Tyrosine的DMEM,2h后加入药物处理细胞,提取300 μl培养液加入TCA 33.3 μl使终浓度为10%,以后每4h取300 μl培养液并加入TCA,最后一次取样后将细胞刮下移入Ep管,离心,沉淀用0.5N纯碱溶解,所有样本取200 μl进行液闪计数。蛋白降解率计算:各时间点cpm值/cpm总和×100%,绘制曲线。
Western blot分析Atrogin-1表达水平CellLyticTMMT细胞裂解液提取总蛋白,考马斯亮蓝检测蛋白浓度,10 % SDS-聚丙烯酰胺凝胶电泳、转膜,加入抗-Atrogin-1(Fbx 一抗);4℃过夜,加入荧光二抗抗山羊或兔IgG(donkey anti-goat/Rabbit IgG)(Invitrogin,Alexa Fluor 680/800),Odyssey激光成像系统(LiCor)扫描,蛋白表达量用内参照GAPDH校正。
Nothern Blot分析Atrogin-1基因沉默采用小分子干扰DNA片段(siRNA)核糖核酸干扰技术,观察DEX作用下C2C12肌管形态及Atrogin-1表达。方法如下:Mafbx-32 siRNA 6 μl 用100 μl siRNA Transfection Media(Invitrogen)稀释,转染肌管24h后,加入DEX 5 μmol/L,继续培养24h,用TRIzol Reagent(invitrogen)提取总RNA,Northern blot检测肌管中Atrogin-1mRNA表达水平。RT-PCR扩增引物制备探针,引物序列Atrogin-1:(forward)5′-GCA AAC ACT GCC ACA TTC TCTC-3′;(reverse)5′-CTT GAG GGG AAA GTG AGA CG-3′;GAPDH(glyceral-dehyde-3-phosphate dehydrogenase)探针购自Ambion。采用随机引物标记试剂盒(Amersham)、α-32P dCTP-标记cDNA 探针,膜置于42℃杂交16h,分别以2×、0.2×SSC/0.1%SDS、去离子水洗膜。薄膜置于暗盒中,压上X光片,置-70℃放射自显影24h。mRNA相对表达量用GAPDH校正。

结 果
地塞米松可引起体外培养肌管萎缩体外培养肌细胞融合形成肌管后,加入绿色荧光蛋白可使肌纤维着色,紫外线下呈绿色,肌管粗壮呈条索状,内见融合的细胞核呈点状强烈荧光,GFP不引起肌纤维性状改变。DEX 0.5 μmol/L作用24h后肌管形态无明显改变,1 μmol/L已引起肌管变细,5 μmol/L则肌纤维明显变细,排列稀疏。DEX 5 μmol/L作用24h后,测量肌纤维直径较正常对照组明显下降[(9.69±2.86)μm DEXvs(18.3±3.57)μm GFP,P<0.01)],提示DEX可引起体外培养肌管萎缩,随DEX剂量增加,肌管逐渐变细(图1 A、B)。
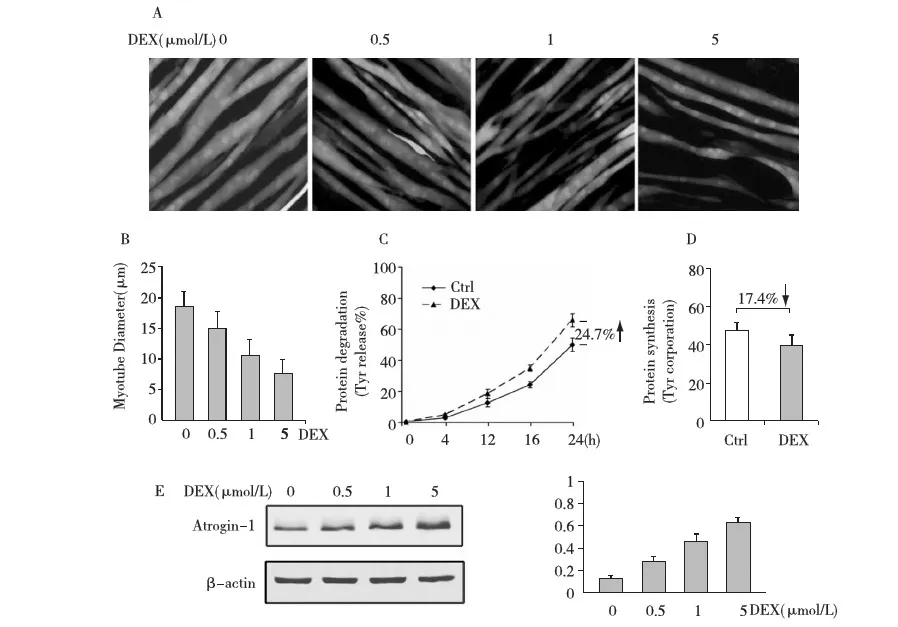
图1 A:培养的C2C12肌细胞经分化后形成肌管,荧光显微镜观察肌管呈条索状,GFP使其着色呈绿色荧光,内见融合的细胞核呈点状强烈荧光;地塞米松(DEX 0~5 μmol/L)作用24h后,DEX > 1 μmol/L可引起肌管萎缩,形态表现为肌纤维变细,排列稀疏(×400); B:DEX(0~5 μmol/L)作用24h后,测量肌管直径逐渐变细; C:DEX 5 μmol/L作用(0~24h)可促进蛋白分解增加,24h时3H-Tyrosine释放率较对照组升高24.7%; D:DEX 5 μmol/L作用24h后蛋白质合成(3H-Tyrosine掺入率)较对照组下降17.4%; E:Western blot显示DEX剂量依赖性地引起Atrogin-1蛋白表达水平升高
地塞米松促使肌管分解代谢和Atroign-1表达升高给予DEX 5 μmol/L作用0~24h,各时间点3H-Tyrosine释放率逐渐增加,16h后与对照值(CTL)比较有统计学意义(表1);24h时3H-Tyrosine释放率升高24.7%;同时间点肌管3H-Tyrosine掺入水平较CTL明显降低[(39.22±6.22)cpm/g DEXvs(47.53±4.32)cpm/g CTL;P=0.047),掺入率下降17.4%。提示DEX可引起蛋白质合成代谢轻度下降,显著促进肌肉蛋白质分解(图1 C、D)。DEX 0~5 μmol/L作用24h,检测Atrogin-1蛋白表达水平升高,以DEX 5 μmol/L作用最显著(图1E)。

表1 地塞米松5 μmol/L作用后各时间点肌管3H-Tyrosine释放率(%)
Atrogin-1基因沉默可改善DEX引起的肌管萎缩应用siRNA技术使Atrogin-1基因沉默,肌管再经DEX 5 μmol/L处理24h后,Northern blot结果提示siRNA可使Atrogin-1 mRNA表达水平显著下降;siRNA对照组Atrogin-1 mRNA表达水平升高约3倍,siRNA-Atrogin-1组仅升高约10%;同时Western blot 结果提示Atrogin-1蛋白表达也显著下降(图2A)。Atrogin-1基因沉默后,再经 DEX 5 μmol/L处理24h,镜下观察肌管形态未见明显改变,SiRNA-Atrogin-1组的肌管直径为[(15.81±3.37)μm DEX)vs(18.63±3.48)μm CTL],而siRNA-CTL组则出现明显肌管萎缩[(8.64±2.41)μm DEXvs(17.36±3.85)μm CTL,P<0.01)]。上述结果提示Atrogin-1基因沉默对DEX引起的肌萎缩具有保护作用。
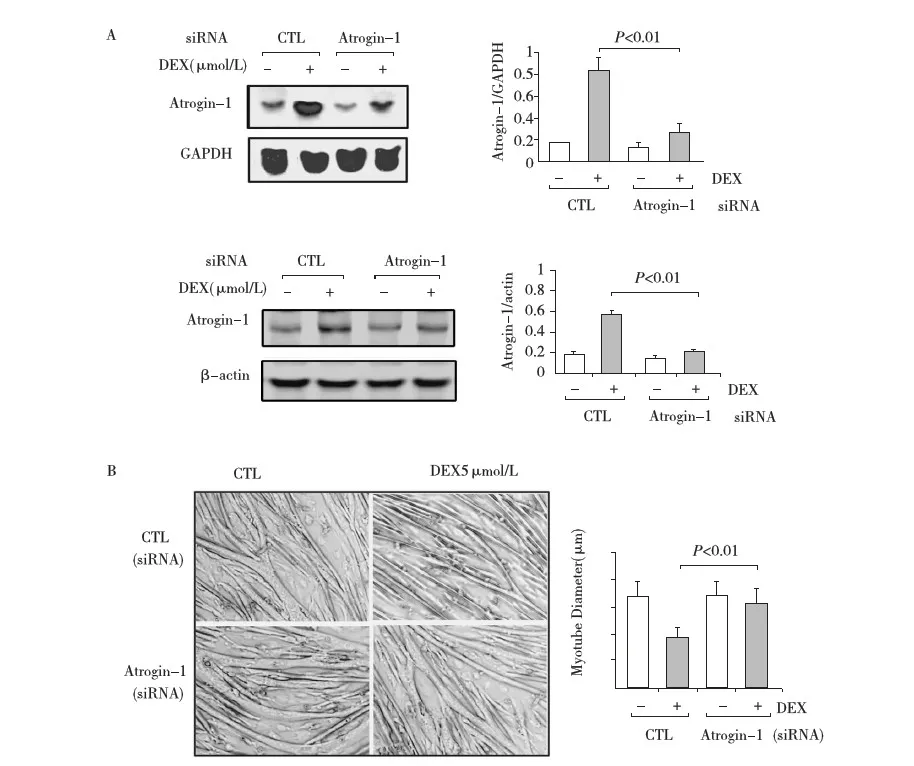
图2 A:Northern blot 显示DEX 5 μmol/L作用24h后,siRNA-对照组的Atrogin-1 mRNA水平升高约3倍,siRNA-Atrogin-1组的Atrogin-1 mRNA 水平较对照组仅升高约10%; Western blot显示靶基因沉默后Atrogin-1蛋白表达水平明显下降;B:光镜下观察肌管形态变化,Atrogin-1基因沉默,再次给予DEX 5 μmol/L作用24h后,未见肌管明显萎缩(×200)
讨 论
近年来研究发现病理条件下内源性GC水平升高可能是促进蛋白质分解的重要因素[5]。May等[6]以NH4Cl造成的代谢性酸中毒大鼠为模型,发现24h尿液中皮质酮水平升高12倍以上;我们既往研究提示尿毒症小鼠模型其内源性GC水平较正常对照组升高13倍以上。本研究采用体外培养骨骼肌细胞,并使之分化为具有肌肉特性的肌管,给予外源性DEX后可以显著刺激肌管萎缩,并影响蛋白质代谢,使肌肉蛋白质合成下降,明显促进蛋白质分解代谢。这表明高水平的GC可降低肌肉蛋白合成,加速蛋白分解。
体内蛋白质分解主要通过3种途径,溶酶体-蛋白酶途径、钙依赖-蛋白酶途径和ATP-泛素-蛋白酶途径,目前确定后者是体内蛋白分解的主要途径。病理状态下肌肉特异性的E3连接酶——Atrogin-1被激活,并通过蛋白酶体直接将骨骼肌蛋白质分解为多肽或氨基酸片段,编码Atrogin-1的基因可能是控制肌肉蛋白分解的关键基因[7]。Mitch等[8]发现切除动物肾上腺或给予GC受体阻滞剂后,可以抑制酸中毒、胰岛素缺乏、饥饿或脓毒血症状态下的肌肉萎缩,若再次给予高于生理剂量的GC后,又可出现UPS激活并引起肌肉萎缩。本研究明确DEX可以剂量依赖性地刺激Atrogin-1蛋白表达,提示GC可直接升高Atrogin-1而引起肌肉萎缩。我们最近对GC引起肌萎缩的机制的研究,揭示GC通过其受体(glucocorticoid receptor,GR)直接干预胰岛素受体底物-1(insulin receptor substrate-1,IRS-1)相关的 PI3K/Akt活性,促进Atrogin-1 mRNA表达,使PI3K/Akt活性下降,引起肌肉萎缩;肌肉特异性GR敲除的转基因小鼠(muscle glucocorticoid receptor knock-out,MGRKO),可抵抗糖尿病引起的Atrogin-1 mRNA表达;我们还发现生理剂量的GC可加剧胰岛素受体敲除小鼠的肌肉蛋白分解,GR激活后可竞争PI3K,这一非基因性的竞争作用损害胰岛素信号,使蛋白分解增加。
目前对骨骼肌消耗性营养不良尚无有效治疗方法,以基因为靶点进行干预是目前最前沿的研究方向。Atrogin-1基因是否专一性地控制骨骼肌萎缩?我们采用RNA干扰技术(RNA interference,RNAi)予以明确。RNAi是一种高效的基因表达阻断技术,它通过体外合成或体内表达含21~23个核苷酸的双链siRNA,对特定基因进行功能封闭或降低表达,以确定靶基因的功能,siRNA 技术也称基因沉默技术(gene silence)。由于 siRNA 具有高度的序列专一性,可以特异地使特定基因沉默,获得功能丧失,它比反义 RNA 技术和同源共抑制法更有效,因此 siRNA 可以作为一种强有力的研究工具[10]。我们对Atrogin-1基因沉默后,再次给予DEX则不引起的肌管萎缩,提示以Atrogin-1基因为治疗靶点,抑制Atrogin-1的表达,对GC作用下的肌萎缩具有保护作用。
综上所述,本研究明确GC通过激活Atrogin-1,引起肌肉细胞蛋白质分解代谢增加,并抑制蛋白合成代谢,从而导致肌肉萎缩;Atrogin-1基因沉默可改善GC引起的肌肉萎缩,Atrogin-1基因可能是逆转肌肉消耗性营养不良的有效靶点。
特别感谢美国Baylor医学院肾脏病实验室胡兆勇博士的指导和帮助!
1 Fouque D,Kalantar-Zadeh K,Kopple J,et al.A proposed nomenclature and diagnostic criteria for protein-energy wasting in acute and chronic kidney disease.Kidney Int,2008,73(4):391-398.
2 Du J,Hu Z,Mitch WE.Molecular mechanisms activating muscle protein degradation in chronic kidney disease and other catabolic conditions.Eur J Clin Invest,2005,35(3):157-163.
3 Waddell DS,Baehr LM,van den Brandt J,et al.The glucocorticoid receptor and FOXO1 synergistically activate the skeletal muscle atrophy-associated MuRF1 gene.Am J Physiol Endocrinol Metab,2008,295(4):E785-E797.
4 王会玲,桂志红,许 烨,等.CKD时高糖皮质激素通过激活 Atrogin-1、MuRF-1引起骨骼肌萎缩.中国中西医结合肾病杂志,2010,11:951-956.
5 Lang CH,Huber D,Frost RA.Burn-induced increase in atrogin-1 and MuRF-1 in skeletal muscle is glucocorticoid independent but downregulated by IGF-I.Am J Physiol Regul Integr Comp Physiol,2007,292(1):R328-R336.
6 May RC,Kelly RA,Mitch WE.Metabolic acidosis stimulates protein degradation in rat muscle by a glucocorticoid-dependent mechanism.J Clin Invest,1986,77(2):614-621.
7 Lecker SH,Jagoe RT,Gilbert A,et al.Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression.FASEB J,2004,18(1):39-51.
8 Mitch WE,Bailey JL,Wang X,et al.Evaluation of signals activating ubiquitin-proteasome proteolysis in a model of muscle wasting.Am J Physiol,1999,276(5 Pt 1):C1132-C1138.
9 Hu Z,Wang H,Lee IH,et al.Endogenous glucocorticoids and impaired insulin signaling are both required to stimulate muscle wasting under pathophysiological conditions in mice.J Clin Invest,2009,119(10):3059-3069.
10 Lagirand-Cantaloube J,Cornille K,Csibi A,et al.Inhibition of atrogin-1/MAFbx mediated MyoD proteolysis prevents skeletal muscle atrophy in vivo.PLoS One,2009,4(3):e4973.
猜你喜欢
中国药理学通报(2022年7期)2022-07-11
肝博士(2022年3期)2022-06-30
中国药理学通报(2022年1期)2022-01-14
海外星云(2021年9期)2021-10-14
当代体育科技(2018年17期)2018-06-11
中国运动医学杂志(2016年3期)2016-07-10
中国运动医学杂志(2016年3期)2016-07-10
武汉轻工大学学报(2015年3期)2015-12-24
医学研究杂志(2015年5期)2015-06-10
实用老年医学(2013年7期)2013-03-11
