
木香药材HPLC指纹图谱的研究
2011-05-26 07:17:52张振秋邓雪男
中成药 2011年6期
李 力, 张振秋, 王 冰, 邓雪男
(辽宁中医药大学药学院,辽宁大连 116600)
木香为菊科植物木香Aucklandiae lappaDecne的干燥根[1],《神农本草经》列为上品[2],其性温,味辛、苦,归脾、胃、大肠、胆、三焦经,具有行气止痛、健脾消食的功效[3-4]。木香中主要含倍半萜内酯类化合物[5-7],其中木香烃内酯和去氢木香内酯的量较高[8],二者的药理活性明显[9]。目前对木香的质量研究中仍未见其HPLC指纹图谱的相关报道,本实验采用HPLC法构建不同来源木香药材的指纹图谱,对10批木香药材进行了相似度评价,为整体控制和评价木香药材的质量提供依据。
1 仪器与试药
1.1 仪器 美国Agilent 1100 Series高效液相色谱仪(四元梯度泵,在线真空脱气机,仪器编号:20041191 DE43607375);G1314-A紫外检测器;AS3120A超声波清洗器;AR2140电子分析天平(上海奥豪斯公司);202-2型干燥箱(上海市实验仪器总厂);METTLER AB135-S十万分之一电子天平(瑞士)。
1.2 试药 木香烃内酯对照品(批号:111524-200503),由中国药品生物制品检定所提供;甲醇为色谱纯,水为纯净水,其余试剂均为分析纯。木香药材样品共10批,分别来源于全国10个地区,经辽宁中医药大学中药鉴定教研室李峰教授鉴定,为菊科植物木香Aucklandiae lappaDecne的干燥根,药材来源见表1。
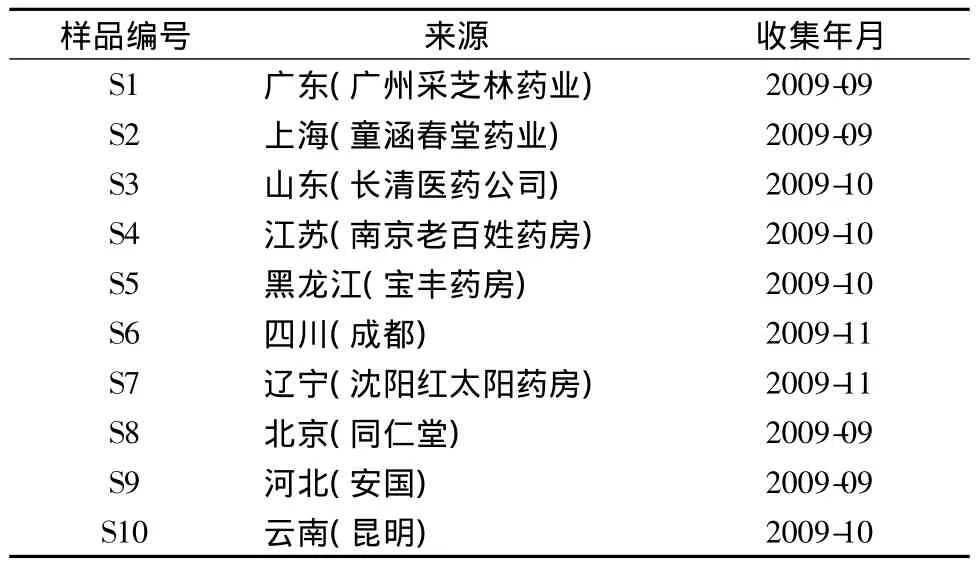
样品编号 来源 收集年月S1 广东(广州采芝林药业)2009-09 S2 上海(童涵春堂药业) 2009-09 S3 山东(长清医药公司) 2009-10 S4 江苏(南京老百姓药房) 2009-10 S5 黑龙江(宝丰药房) 2009-10 S6 四川(成都) 2009-11 S7 辽宁(沈阳红太阳药房) 2009-11 S8 北京(同仁堂) 2009-09 S9 河北(安国) 2009-09 S10 云南(昆明)2009-10
2 方法与结果
2.1 色谱条件 色谱柱Phenomsil ODS(250 mm×4.6 mm,5 μm),流动相:甲醇(A)-水(B)梯度洗脱,体积流量1.0 mL/min,柱温30℃,检测波长254 nm,进样量10 μL,梯度洗脱程序见表 2,所有组分均在70 min内被洗脱。

表2 流动相梯度洗脱程序Tab.2The schedules of mobile phase gradient elution
2.2 对照品溶液的制备 精密称取木香烃内酯对照品1.98 mg,置25 mL量瓶中,加甲醇稀释至刻度,摇匀,即得浓度为0.079 2 mg/mL的对照品溶液。
2.3 供试品溶液的制备 取木香样品粗粉0.5 g,精密称定,置具塞锥形瓶中,精密加甲醇50 mL,密塞,摇匀,称定质量,放置过夜,超声处理(功率250 W,频率50 kHz)30 min,取出,放冷,再称定质量,用甲醇补足减失的质量,摇匀,滤过,取续滤液,即得供试品溶液。
2.4 方法学考察
2.4.1 精密度试验 取同一木香药材供试品溶液(样品号10),按2.1项下色谱条件进行测定,连续进样6次,考察色谱峰相对保留时间和相对峰面积的一致性,结果其共有峰的相对保留时间和相对峰面积 RSD 分别为 0.02% ~0.23% 和 0.22% ~1.9%,说明仪器精密度良好,符合指纹图谱测定的要求[10-11]。
2.4.2 稳定性试验 取同一木香药材供试品溶液(样品号10),按2.1 项下色谱条件分别在0、2、4、8、12、24 h进行测定,结果其共有峰的相对保留时间和相对峰面积RSD分别为0.02% ~2.6%和1.5%~2.7%,说明供试品在24 h内基本稳定。
2.4.3 重复性试验 精密称取同一批木香药材粗粉(样品号10)6份,按2.3项下供试品溶液的处理方法平行制得6份供试品溶液。按2.1项下色谱条件进行测定,结果其共有峰的相对保留时间和相对峰面积的RSD均小于3%,表明重复性好。
2.5 样品指纹图谱测定 分别取来自10个不同来源的木香样品,按2.3项下供试品溶液制备方法得供试品溶液。按2.1项下色谱条件进行测定,记录HPLC图。比较不同来源样品的色谱图,其中13个色谱峰为10批样品的共有峰,确定为共有指纹峰,其中9号峰为木香烃内酯色谱峰,选作参照峰,设其保留时间和峰面积分别为1,其他各共有峰的保留时间和峰面积分别与参照峰的保留时间和峰面积相比,计算各样品指纹图谱中共有峰的相对保留时间和相对峰面积,结果分别见图 1、2、3,表 3、4。

图1 木香烃内酯对照品HPLC色谱图Fig.1 HPLC chromatogram of costunolide
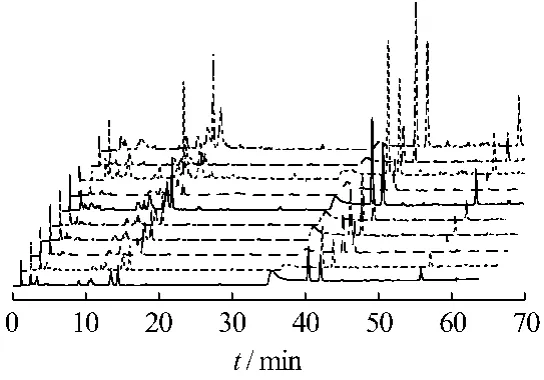
图2 10个木香样品的HPLC指纹图谱Fig.2 HPLC fingerprint of 10 samples

图3 木香药材的HPLC指纹共有模式Fig.3 HPLC fingerprint mutual mode of Aucklandiae Radix
2.6 指纹图谱的相似度计算[12]运用国家药典委员会推荐的中药色谱指纹图谱相似度评价系统2004A版对10批木香药材的指纹图谱进行了相似度分析,将色谱工作站的数据导入中药指纹图谱相似度计算软件,选定上述13个特征峰进行谱峰匹配,通过中位数矢量计算得出木香样品指纹图谱的共有模式,并依此共有模式为标准,进行整体相似度评价。从表5可以看出,10批木香药材的相似度均大于0.90,说明各地区的样品色谱模式相似,其化学成分一致性较好。相似度评价结果见表5。
3 讨论
3.1 样品处理方法的考察 考察了超声、回流、冷浸等提取方法,其中超声提取的样品图谱杂质干扰少、色谱峰基线分离好,故选用超声提取法提取。提取溶剂分别用甲醇、乙醇、三氯甲烷、乙酸乙酯超声提取30 min,结果表明甲醇提取杂质少、提取完全,故选择甲醇作为提取溶剂。分别超声提取15 min、30 min、45 min,结果随着提取时间的延长,色谱峰的面积有所增大,超声提取30 min时提取较完全,超声45 min提取所得的峰面积有所减小,所以提取时间选择超声30 min。

表3 10批木香药材的相对保留时间Tab.3 Relative retention time of 10 samples

表4 10批木香药材的相对峰面积Tab.4 Relative peak areas of 10 samples

表5 木香样品指纹图谱相似度分析结果Tab.5 Similarity analysis results of Aucklandiae Radix
3.2 色谱条件的选择 试验过程中选择了多种流动相系统(等度洗脱,梯度洗脱):甲醇-水、乙腈-水、甲醇-磷酸水(0.05%)等流动相系统洗脱,并对梯度洗脱程序进行优化,结果表明以甲醇-水系统,实验所确定的梯度程序使各色谱峰达到基线分离,峰型对称,分离较好,保留时间适中。实验比较了20℃、30℃、40℃ 3个不同柱温,发现随柱温上升各色谱峰保留时间提前,在40 min后出现的色谱峰变化幅度较大,在30℃时检测相对保留时间稳定,因此选择30℃为指纹图谱的检测柱温。
3.3 从不同来源木香药材的指纹图谱中可以看出,各特征峰的相对保留时间相似度较高,可用于木香药材的图谱鉴别,但峰面积比值不尽相同,差异较大,说明木香药材的质量受地理环境和气候条件影响较大,因此木香药材生产必须固定地区,才能确保其质量的稳定可控。
3.4 10批木香药材指纹图谱的相似度均在0.90以上,说明按本方法进行指纹图谱分析,其相似性良好。所建立的标准对照色谱指纹图谱,峰的数目、强度及分离度均较理想,满足了指纹图谱的要求,并且通过方法学考查试验验证,本试验具有较好的精密度、稳定性和重复性,所以本方法可对木香药材进行质量控制。
[1]国家药典委员会.中华人民共和国药典:2010年版一部[S].北京:中国医药科技出版社,2010:57-58.
[2]魏·吴 普,清·孙星衍,孙冯冀辑.神农本草经[M].北京:人民卫生出版社,1997:186.
[3]林明峡.木香的药理及临床研究概况[J].中医药信息,2005,22(3):18-19.
[4]江苏新医学院.中药大辞典:上册[M].上海:上海科技出版社,1979:353.
[5]邱 琴.GC-MS法测定木香挥发油化学成分[J].理化检验-化学分册,2001,37(8):346.
[6]Singh I P,Talwar K K,Chhabra B R,et al.A biologically active guaianolide from Saussurea lappa[J].Photochemistry,1992,31(7):2529-2531.
[7]Talwar K K,Singh I P,Kalsi P S.A sesquiterpenoid with plant growth regulatory activity from S aussurea lappa[J].Photochemistry,1992,31(1):336-338.
[8]徐 宇,方鲁延,谈 红,等.HPLC法测定木香中去氢木香内酯的含量[J].中草药,2004,35(12):1416-1417.
[9]王永兵,王 强,毛福林,等.木香的药效学研究[J].中国药科大学学报,2001,32(2):146-148.
[10]王 瑾,胡臻华.中药指纹图谱的研究概述[J].解放军药学学报,2004,20(3):214-216.
[11]侯小平,何新新,苏薇薇.中药指纹图谱质量控制技术[J].中药材,2002,24(5):370-371.
[12]陈思妮,张振秋.广金钱草HPLC指纹图谱的研究[J].中成药,2008,30(9):1249-1252.
猜你喜欢
CHINA TODAY(2022年8期)2022-08-03 07:41:34
小小说月刊(2022年14期)2022-07-18 07:37:02
小哥白尼(趣味科学)(2021年11期)2021-02-28 08:35:00
小天使·一年级语数英综合(2020年10期)2020-12-16 02:57:11
天津中医药(2020年5期)2020-06-01 12:16:14
现代装饰(2017年11期)2017-05-25 02:15:19
中成药(2017年4期)2017-05-17 06:09:27
中国民族医药杂志(2016年6期)2016-05-09 08:52:57
自动化学报(2016年8期)2016-04-16 03:39:00
青少年科技博览(中学版)(2015年7期)2015-08-12 18:50:24
