
环孢菌素A对脂多糖诱导的小鼠急性肺损伤的保护作用*
2011-05-24 16:50胡俊锋夏雪梅李殿明陈余清
中国应用生理学杂志 2011年1期
胡俊锋,夏雪梅,李殿明,张 永,陈余清
(蚌埠医学院第一附属医院呼吸内科,安徽 蚌埠 233004)
急性肺损伤(acute lung injury,ALI)是全身炎症导致多器官功能障碍综合征的肺部表现,严重阶段为急性呼吸窘迫综合征(acute respiratory distress syndrome,ARDS),临床表现为呼吸频数和呼吸窘迫,顽固性低氧血症,后期多并发多器官功能障碍。尽管临床上对ALI采取了多种综合治疗手段,但病死率仍然较高。因此探索治疗新靶点,寻找和研发新的治疗药物是研究者和临床医生的目标。
脂多糖(lipopolysaccharide,LPS)是诱导ALI动物模型最常用的药物之一,其主要成分内毒素可导致肺组织细胞损伤,使细胞结构和功能发生病理改变。线粒体是能量代谢的主要场所。线粒体渗透性转换孔道 (mitochondrial permeability transition pore,mitoPTP)是存在于线粒体内外膜上的一非特异性孔道,其开放是各种器官和细胞损伤或死亡的共同通路[1]。环孢菌素A(cyclosporine A,CsA)可抑制线粒体渗透性转换孔道的开放而发挥保护作用。文献报道LPS诱导的急性肺损伤可导致线粒体膜肿胀,脂质过氧化增加,膜流动性降低,线粒体活力下降及ATP生成减少等[2]。LPS诱导的ALI是否通过改变线粒体上的重要作用环节——mitoPTP的开放状态产生效应?使用该孔道的抑制剂CsA是否可发挥抗LPS诱导的急性肺损伤作用?目前报道甚少。本研究着重观察mitoPTP抑制剂CsA对LPS诱导的小鼠ALI模型是否可发挥保护作用,分析其可能的线粒体机制,为揭示ALI的发病机制提供理论依据,并为临床防治提供新思路。
1 材料与方法
1.1 动物和分组
清洁级健康雄性ICR品系小鼠,120只,体质量20~30g,由蚌埠医学院实验动物中心提供。
将小鼠随机分为5组(n=24):对照组、LPS组、地塞米松组、CsA组和CsA+苍术苷(atractyloside,Atr)组。腹腔注射100g/L水合氯醛麻醉小鼠后,小心钝性分离并暴露气管,气管内缓慢滴入LPS 4 mg/kg为LPS组,对照组给予相同容积的生理盐水(2 ml/kg),地塞米松组、CsA组分别于造模后30min尾静脉注射地塞米松5 mg/kg和mitoPTP抑制剂CsA 5 mg/kg。CsA+Atr组于造模后30min尾静脉同时注射mitoPTP抑制剂CsA 5 mg/kg和开放剂Atr 5 mg/kg。
1.2 药品与试剂
脂多糖(lipopolysaccharide,LPS,大肠埃希菌O127:B8)、环孢菌素A(cyclosporine A,CsA)、苍术苷(atractyloside,Atr)、伊文斯蓝(Evans blue)为 Sigma 公司产品;地塞米松(dexamethasone,Dx)购于浙江仙琚制药公司。小鼠肿瘤坏死因子-α(tumor necrosis factor-α,TNF-α)测定试剂盒购于武汉博士德公司。其余为国产分析纯。
1.3 小鼠急性肺损伤实验模型复制
实验动物适应环境后,复制LPS气管内滴入诱导小鼠ALI模型。腹腔注射100g/L水合氯醛麻醉小鼠后,小心钝性分离并暴露气管,气管内滴入LPS 4 mg/kg,6 h后麻醉小鼠,股动脉放血处死,观察各项指标[3]。
1.4 支气管肺泡灌洗液LDH的测定
造模6 h后,每组随机取8只小鼠,麻醉后股动脉放血处死,暴露气管行气管插管,用PBS液1.5 ml分3次进行支气管肺泡灌洗术,回收率达80%。采用分光光度法测定支气管肺泡灌洗液(bronchoalveolar lavage fluid,BALF)中乳酸脱氢酶(lacate dehydrogenlase,LDH)的活性反映肺组织损伤程度。原理为LDH催化丙酮酸盐还原生成乳酸,同时NADH被氧化成NAD+,引起340nm吸光度的下降,其下降速率与样本中LDH活力成正比,通过吸光度变化率计算LDH活性。
1.5 肺组织匀浆液TNF-α活性和肺水含量的测定
各组中随机取另外8只小鼠,取其左肺,肺组织匀浆液,采用酶联免疫吸附法测定TNF-α含量。
右肺滤纸吸干,称取肺湿重,并随即置于60℃烘箱烘烤48 h后称干重,计算肺组织湿重/干重比反映肺水含量。
1.6 肺毛细血管通透性测定
各组中取剩余的8只小鼠,LPS滴入气管5.5 h后,以10g/L伊文斯蓝5 ml/kg尾静脉注射,30min后,麻醉小鼠,打开胸腔暴露心肺,剪除心脏与肺门组织,滤纸吸干血液和肺周围水分,精密称取肺组织,用手术剪将肺组织剪成均匀小块,置试管内,每l00mg肺组织加入丙酮-生理盐水溶液(7∶3,v/v)3 ml,置室温(25℃)24h,然后1500r/min离心10min,取上清用分光光度计620nm比色测定浸出液吸光度反映湿肺中伊文斯蓝的含量。
1.7 统计学分析
2 结果
2.1 小鼠的一般状况
对照组小鼠活泼好动,呼吸均匀。小鼠气管内滴入LPS后,呼吸频率明显加快,随时间的延长,小鼠精神差,活动减少。给予地塞米松和CsA,小鼠精神状态好转,呼吸频率减慢,提示该两种药物具有改善作用。
2.2 小鼠支气管肺泡灌洗液中LDH的变化
与对照组相比,LPS组BALF中LDH显著升高。CsA和地塞米松可明显降低BALF中LDH的增加(P<0.01),CsA+Atr组与LPS组无明显区别(图1)。

Fig.1 Effect of cyclosporine A(CsA,5 mg/kg),dexamethasone(Dx,5 mg/kg)and atractyloside(Atr,5 mg/kg)on LDH activity in bronchoalveolar lavage fluidof LPS-induced acute lung injury mice(,n=8)
2.3 小鼠肺组织匀浆液中TNF-α浓度的变化
与对照组相比,LPS组肺组织匀浆液中的TNF-α浓度明显升高;与LPS组相比,地塞米松和CsA组肺组织匀浆液中的TNF-α浓度明显降低(P<0.01),CsA+Atr组TNF-α浓度与LPS组相比无明显改变(图2)。

Fig.2 Effect of cyclosporin A(CsA,5 mg/kg),dexamethasone(Dx,5 mg/kg)and atractyloside(Atr,5 mg/kg)on tumor necrosis factor-α(TNF-α)level in lung tissue homogenate of LPS-induced acute lung injury mice(,n=8)
2.4 小鼠肺水含量和肺毛细血管通透性的变化
与对照组相比,LPS组小鼠肺组织湿重/干重比明显升高。伊文斯蓝渗出结果表明,LPS组620nm吸光度明显高于对照组(P<0.01),提示肺毛细血管伊文斯蓝渗出增多。与LPS组相比,CsA组肺水含量降低,620nm吸光度降低,提示伊文斯蓝渗出减少。CsA+Atr组无明显变化(表1)。
3 讨论
LPS诱导的小鼠急性肺损伤动物模型可模拟临床病人内毒素升高引起的急性肺损伤或急性呼吸窘迫综合症的形态和功能变化。对于急性肺损伤,临床治疗应用糖皮质激素可控制肺损伤伴发的炎症和肺水肿。本实验中我们通过不同指标观察到糖皮质激素地塞米松可发挥抗LPS诱导的肺损伤的作用,与以往报道一致。但长期使用激素治疗常伴随神经肌变等较严重的副反应[4]。因此寻求新的、科学的、有效的治疗手段有利于更广泛地临床应用。
本研究结果显示线粒体渗透性转换孔道mitoPTP抑制剂CsA可使LPS诱导的小鼠急性肺损伤的BALF中LDH释放减少,肺组织匀浆液中TNF-α浓度降低,同时降低了肺组织湿重/干重比,肺组织伊文斯蓝渗出减少,表明肺组织通透性降低。CsA组各项指标与地塞米松组无明显区别,提示CsA可发挥抗LPS诱导的急性肺损伤的作用。mitoPTP开放剂Atr取消了CsA的保护作用,提示CsA的保护作用可能与其抑制mitoPTP开放有关。
Tab.1 Effect of cyclosporin A(CsA),dexamethasone(Dx)and atractyloside(Atr)on lung wet weight/dry weight ratio and lung permeability of LPS-induced acute lung injury mice(,n=8)
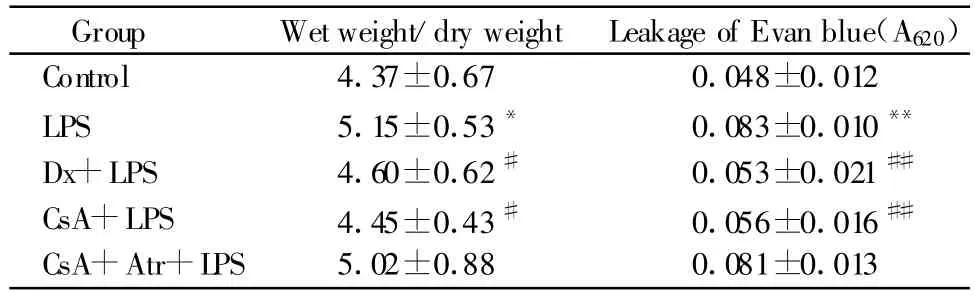
Tab.1 Effect of cyclosporin A(CsA),dexamethasone(Dx)and atractyloside(Atr)on lung wet weight/dry weight ratio and lung permeability of LPS-induced acute lung injury mice(,n=8)
LPS:Lipopolysaccharide group;Dx+LPS:Dexamethasone+lipopolysaccharide group;CsA+LPS:cyclosporine A(inhibitor of mitoPTP)+lipopolysaccharide group;CsA+Atr+LPS:Cyclosporine A+atractyloside(opener of mitoPTP)+lipopolysaccharide group*P<0.05,**P<0.01 vs control group;#P<0.05,##P <0.01 vs LPS group
Group Wet weight/dry weight Leakage of Evan blue(A620)Control 4.37±0.67 0.048±0.012 LPS 5.15±0.53* 0.083±0.010**Dx+LPS 4.60±0.62# 0.053±0.021##CsA+LPS 4.45±0.43# 0.056±0.016##CsA+Atr+LPS 5.02±0.88 0.081±0.013
LPS可诱导全身性损伤,引起各脏器线粒体功能改变,导致呼吸链功能障碍,破坏线粒体结构、引起mitoPTP开放等,最终引起脏器功能障碍[5]。mitoPTP被认为是各种损伤(应激、缺氧复氧、钙超载等)的终末效应器之一。CsA能抑制mitoPTP开放,减弱细胞凋亡和坏死的发生[1]。已有报道,CsA可抑制LPS诱导的心脏损伤、肝脏损伤、回肠损伤等,其机制与其抑制mitoPTP的开放有关[6-8]。既然各脏器的损伤均与线粒体机制有关,我们提出,LPS引起肺损伤是否与肺线粒体损伤有关。关于线粒体与肺损伤之间的关系,目前报道不多。缺氧诱导的肺损伤中,mitoPTP的开放参与其中[9]。慢性阻塞性肺病患者,其骨骼肌和呼吸肌的线粒体出现病变,线粒体肿胀,细胞色素C释放增多。盲肠穿孔诱导的小鼠败血症模型中,CsA降低了BALF中的蛋白含量,肺组织MPO释放减少,提示CsA可减轻败血症引起的肺组织损伤。本研究采用了气道内滴入LPS减少全身反应的干预,更直接地观察肺损伤变化。实验结果提示mitoPTP的开放亦可能参与气道内滴入LPS诱导的急性肺损伤,进一步提示线粒体机制可能是各重要脏器损伤的共同靶点。
许多研究证明气道内滴入LPS可诱导一系列炎症细胞释放多种炎症介质如TNF-α等,导致炎症免疫网络的激活,从而引起急性炎症反应。TNF-α在线粒体功能障碍发生中可能发挥了极其重要的作用。血清中TNF-α浓度增高可引起活性氧产生增加,清除减少,导致氧化应激引起线粒体损伤。TNF-α与细胞膜上相应受体结合可使Bax/Bcl-2比值增加,促进mitoPTP的开放和诱导线粒体依赖途径凋亡的发生。已报道TNF-α等可引起mitoPTP构象改变,促进孔道的开放,引起下游凋亡的发生[10]。本实验中,线粒体渗透性转换孔道抑制剂CsA减弱了肺组织匀浆液中TNF-α的释放,提示CsA可能通过减少炎症因子的释放,进一步抑制mitoPTP的开放,降低LPS诱导的ALI的程度,其具体机制还有待进一步证实。
[1]Halestrap A P,Clarke S J,Javadov S A.Mitochondrial permeability transition pore opening during myocardial reperfusion-a target for cardioprotection[J].Cardiovasc Res,2004,61(3):372-385.
[2]尚 涛,张建新,李兰芳,等.氨基胍对内毒素性肺损伤大鼠肺脏线粒体损伤的影响[J].中国药理学通报,2007,23(7):870-874.
[3]Tang H F,Lu J J,Tang J F,et al.Action of a novel PDE4 inhibitor ZL-n-91 on lipopolysaccharide-induced acute lung injury[J].Int Immunopharmacol,2010,10(4):406-411.
[4]Steinberg K P,Hudson L D,Goodman R B,et al.Efficacy and safety of corticosteroids for persistent acute respiratory distress syndrome[J].N Engl J Med,2006,354(16):1671-1684.
[5]Crouser E D.Mitochondrial dysfunction in septic shock and multiple organ dysfunction syndrome[J].Mitochondrion,2004,4(5-6):729-741.
[6]Zhuge J,Cederbaum A I.Inhibition of the mitochondrialpermeability transition by cyclosporin A prevents pyrazole plus lipopolysaccharide-induced liver injury in mice[J].Free Radic Biol Med,2009,46(3):406-413.
[7]Larche J,Lancel S,Hassoun SM,et al.Inhibition of mitochondrial permeability transition prevents sepsis-induced myocardialdysfunction and mortality[J].J Am Coll Cardiol,2006,48(2):377-385.
[8]Crouser E D,Julian M W,Joshi M S,et al.Cyclosporin A ameliorates mitochondrial ultrastructural injury in the ileum during acute endotoxemia[J].Crit Care Med,2002,30(12):2722-2728.
[9]Pagano A,Donati Y,Métrailler I,et al.Mitochondrial cytochrome C release is a key event in hyperoxia-induced lung injury:protection by cyclosporin A[J].Am J Physiol Lung Cell Mol Physiol,2004,286(2):L275-283.
[10]Tafani M,Schneider T G,Pastorino J G,et al.Cytochrome c-dependent activation of caspase-3 by tumor necrosis factor requires induction of the mitochondrial permeability transition[J].Am J Pathol,2000,156(6):2111-2121.
猜你喜欢
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中国临床医学影像杂志(2022年5期)2022-07-26
水泵技术(2022年2期)2022-06-16
内燃机与动力装置(2022年1期)2022-03-21
海洋通报(2021年1期)2021-07-23
哈尔滨工程大学学报(2021年2期)2021-03-19
生物学通报(2021年4期)2021-03-16
湖北农机化(2020年4期)2020-07-24
中华建设(2019年7期)2019-08-27
中医眼耳鼻喉杂志(2019年3期)2019-04-13
