
亚甲基蓝比色法测定海参不同组织酸性黏多糖含量
2011-03-14 06:06王泽文冷凯良翟毓秀孙伟红邢丽红苗钧魁
海洋科学 2011年3期
王泽文,冷凯良翟毓秀孙伟红邢丽红苗钧魁
(1.中国水产科学研究院 黄海水产研究所,山东 青岛 266071;2.中国海洋大学 食品科学与工程学院,山东青岛 266100)
海参(Sea cucumber)属于棘皮动物门(Echinodermata)、海参纲(Holothuroidea)[1]。其肉质酥脆、香酥滑润、高蛋白质、低脂肪、营养丰富、味道鲜美,是“海味八珍”之一的佳肴和滋补保健品,素有“滋补靠海参、抢救靠人参”的说法。
海参酸性黏多糖是海参特有的重要活性成分,目前已经从刺参、花刺参、玉足海参、梅花参等海参体内分离鉴定出酸性黏多糖,其含量约占干海参总有机物的 6%以上[2]。海参酸性黏多糖(Acid mucopolysaccharide)又称为糖胺聚糖(Glycosaminoglycans),是由氨基半乳糖、葡萄糖醛酸、岩藻糖等组成的分支杂多糖,海参酸性黏多糖中硫酸酯基的含量非常高。近20年来,国内外学者对海参黏多糖及其异构物的药理作用进行了大量研究,证实它具有抗凝血、降血脂、降低血黏度、抗肿瘤以及提高免疫功能等多种活性[3-4]。海参中的酸性黏多糖组成较复杂,如何准确测定其含量一直被人们所关注。目前海参酸性黏多糖含量测定方法主要有薄层色谱法[5]、分光光度法[6],本实验利用亚甲基蓝与海参多糖的异染效应,建立并完善了亚甲基蓝显色分光光度法测定多糖含量的技术。目前国内外关于海参酸性黏多糖的研究多局限于同类或不同种类的整只海参中黏多糖含量研究,对海参不同组织的黏多糖含量测定至今鲜有报道。由于海参肠、卵及体壁是海参体中具有经济价值的主要部位,因此利用建立的方法测定了海参不同组织酸性黏多糖含量,为海参黏多糖的利用提供了数据支持和理论指导。
1 材料与方法
1.1 材料
1.1.1 仪器与设备
LAMBDA750紫外可见分光光度计:美国珀金埃尔默公司;BT224S电子分析天平:法国沙多利斯公司;CR22G型离心机:日本日立公司;CS101-1EB烘箱:四川四达公司。
1.1.2 试剂
木瓜蛋白酶(上海蓝季科技公司,酶活为 100万U/g) ;半胱氨酸(上海惠兴生化试剂公司,纯度大于98.5%);乙酸钠,EDTA、磷酸二氢钾和磷酸氢二钠、亚甲基蓝溶液(0.4mmol/L)、D-甘露糖(Man)购自天津博迪化工有限公司;岩藻糖(Fuc)、D-葡萄糖(Glu)、D-半乳糖(Gla) 购自国药集团化学试剂有限公司。
磷酸缓冲液(pH为5.0):称取磷酸二氢钾1.701 2 g,溶于500 mL水中。称取磷酸氢二钠0.222 6 g溶于50 mL水中,取磷酸二氢钾溶液490 mL,磷酸氢二钠溶液10 mL混合,此缓冲液pH为5.0。
乙酸缓冲液(pH 为 6.0):取乙酸钠 54.6 g,加1 mol/L乙酸溶液20 mL溶解后,加水稀释至500 mL。
多糖提取液:用乙酸缓冲液配制成浓度为 5 mmol/L半胱氨酸和EDTA的混合溶液,并加入木瓜蛋白酶(500 mL加1.67 g,准确到0.001 g)。
酸性黏多糖标准品:Fluka公司提供,CAS号19072-19-9,硫酸酯基含量为9%~10%。以上试剂均为分析纯级或优级纯,所用水为去离子水。
1.1.3 样品
海参肠、海参卵、海参体壁冻干粉均由本实验室自制,该海参品种为刺参(Stichopus japonicus)。
1.2 方法
1.2.1 异染配合物最大吸收波长的确定
取1mL亚甲基蓝溶液于 10mL容量瓶中,用磷酸缓冲液定容至刻度,然后分别取0.2 g/L标准溶液0.4、0.8、1.0 mL于瓶中,加入1 mL亚甲基蓝溶液,用磷酸缓冲液定容至10 mL。用紫外可见分光光度计从 350~750 nm进行扫描,记取不同波长下溶液的吸光值。
1.2.2 标准曲线的绘制
精密称取 105℃干燥至恒重的海参多糖标准品,用磷酸缓冲液配置成0.2 g/L的标准溶液,分别取配好的多糖标液0、0.2、0.4、0.6、0.8、1.0 mL,置于10 mL容量瓶中,向其中加入 1 mL亚甲基蓝溶液,用磷酸缓冲溶液定容至刻度,配制成一系列浓度的标准溶液。未加标准的溶液作为空白参比样。以多糖含量为横坐标,吸光值为纵坐标,在564 nm波长下测定吸光值,绘制标准曲线。
1.2.3 样品的处理
取海参各组织样品,于冷冻干燥机中冻干,准确称取 1 g冻干后样品,于 50 mL离心管中,加入30 mL提取液,于 60℃下酶解,振荡提取 24 h,4 000 r/min离心5 min,收集上清液,用乙酸缓冲液定容到50 mL,待测定。
1.2.4 样品检测
准确移取 1.2.4得到的酶解液 0.1 mL,放于10 mL容量瓶中,加入1 mL亚甲基蓝溶液,用磷酸缓冲液定容至刻度,在564 nm波长下测定各溶液吸光值,通过标准曲线得到溶液中多糖浓度,按照下式计算样品中海参酸性黏多糖的含量。
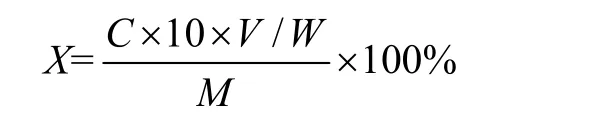
式中,X为样品中酸性黏多糖含量(%);C为样品中测定的酸性黏多糖浓度;V为样品溶液定容体积;W为样品测试取样体积;M为取样质量;10为测定溶液定容体积。
1.3 海参酸性黏多糖经酸水解后液相色谱测定的色谱条件
Eclipse XDB-C18柱 250 mm×4.6 mm,5µm;柱温:30℃;流速:1.0 mL/min,紫外检测波长:250 nm;进样体积:10 µL;流动相:溶剂 A,15%(V/V)乙腈+50 mmol/L磷酸缓冲液(KH2PO4-NaOH,pH 6.0),溶剂B,40%(V/V)乙腈+50 mmol/L磷酸缓冲液(KH2PO4-NaOH,pH 6.0);二元梯度洗脱程序:时间梯度0→9 min→15 min,相应的浓度梯度为100%→90%→45% (溶剂 A)。
2 结果与分析
2.1 方法原理
亚甲基蓝是一种醌亚胺类阳离子染色剂,在酸性介质中,亚甲基蓝的杂环氮原子因质子化而带正电以阳离子状态存在[7],因此能够与负离子亲水胶体相互作用,形成可溶于水的异染变色配合物,这个配合物导致染料颜色发生变化,从蓝色(最大吸收波长 610~664nm)到紫色(最大吸收波长 520~560nm)。海参多糖是由岩藻糖等单糖和有机硫酸酯组成的线性聚阴离子高分子,在酸性介质中,以阴离子状态存在,与亚甲基蓝可发生特异性异染现象,由于亚甲基蓝和聚阴离子间的相互作用是可逆的,且二者之间的结合比率为1:1[8],因此可依据此原理建立海参酸性黏多糖含量测定的亚甲基蓝显色分光光度法。
2.2 异染配合物最大吸收波长的确定
通过图1中 A可以看出,亚甲基蓝水溶液在608 nm和664 nm处有明显吸收峰,当加入不同浓度的酸性黏多糖标液后(图B、C和D),564 nm处的吸光值随着多糖浓度的增加,呈现明显增大的趋势,而 608 nm和664 nm处吸收值呈现明显减少的趋势。这说明海参酸性黏多糖与亚甲基蓝发生了特异性异染效应,生成了一种易溶于水的异染变色配合物,而564 nm正是该异染变色配合物的最大吸收峰,这与Soedjak等[9]报道的结果基本相同,因此可以根据不同浓度酸性黏多糖与次甲基蓝异染后配合物最大吸收波长吸光度值的变化来对样品中多糖含量进行测定。
2.3 样品水解方式的选择
目前多糖的提取主要采用酶水解[10]和酸水解方式(6 mol/L盐酸于105℃充氮封管水解4 h),实验中分别采用两种水解方式对样品进行处理,并利用分光光度法进行多糖样品含量测定。
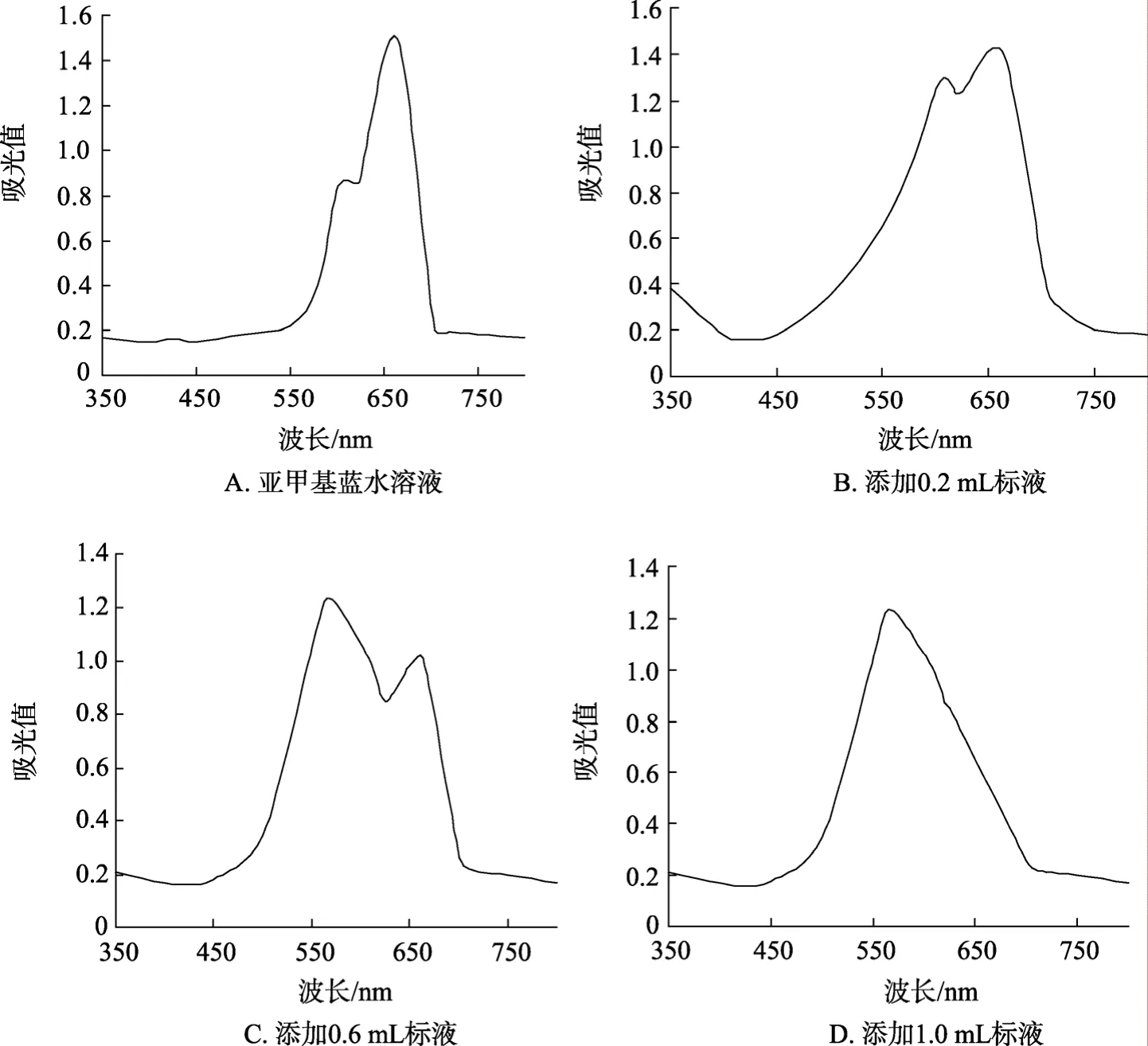
图1 不同浓度标液与亚甲基蓝异染后吸光值变化趋势Fig.1 Changes of absorptions during the mixing of standard solutions and methylene blue
通过图2可以看出,采用酸水解得到的样品与亚甲基蓝呈色反应后,经分光光度计测得的吸光值要明显低于采用酶水解的样品,这主要是由于实验中采用的是木瓜蛋白酶,该酶专一性水解蛋白,而对于其他多糖类物质没有切除作用,因此溶液中剩余的糖类物质是以多糖大分子的形式存在,当加入充足的亚甲基蓝后,多糖大分子与其发生异染反应,生成了在564 nm处有最大吸收的异染配合物。当采用酸水解时,由于盐酸的水解无选择性,致使样品中的多糖被部分水解,经高效液相色谱法测定酸水解产物中含有岩藻糖、甘露糖、半乳糖、葡萄糖等单糖及葡萄糖醛酸(图3),分别将岩藻糖、甘露糖、葡萄糖、半乳糖与亚甲基蓝染料进行混合反应,并采用分光光度法测定其吸光值,发现添加各单糖后样品吸光值与空白样品无异,而葡萄糖醛酸其含量过低(按面积归一化法测定含量约为 3.5%)因此对吸光度值的影响可以忽略,由此可以推断以上几种单糖不能与亚甲基蓝染料发生异染效应,所以经过酸水解后样品中能够与亚甲基蓝产生配合物的物质比酶水解的样品中减少,相应地其在564 nm处吸光值也就发生了降低,这也就与图1得到的结果相吻合。通过酸性黏多糖水解方式的讨论可以发现,蛋白酶水解对于亚甲基蓝分光光度法最适用的。
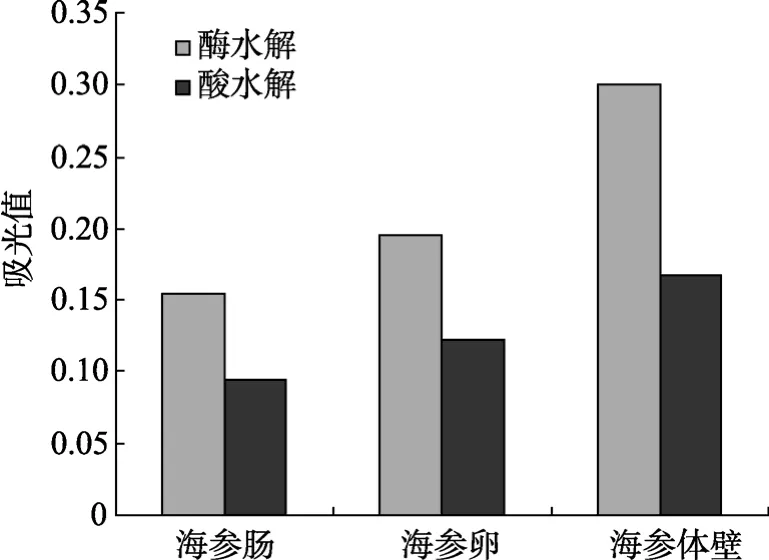
图2 不同水解方式对样品吸光值影响Fig.2 Effects of different ways of hydrolysis on the absorption values of the reaction products
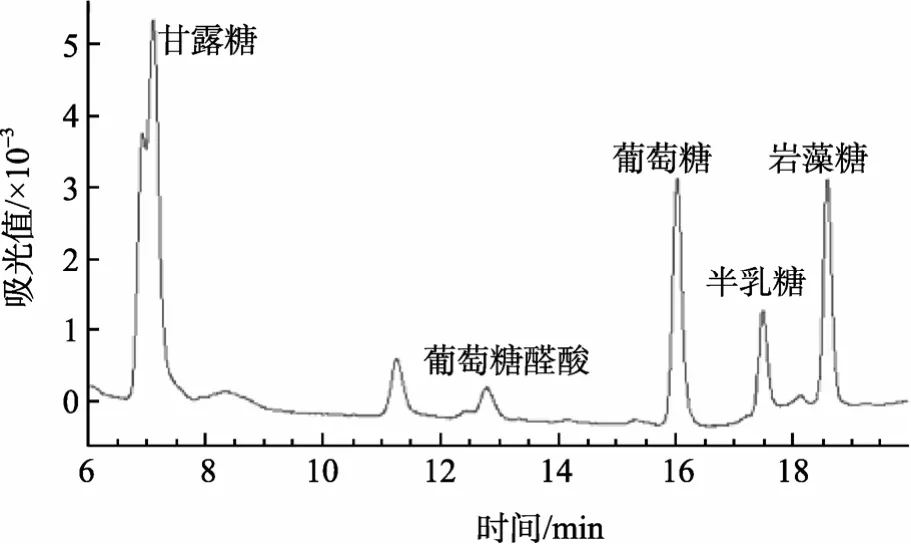
图3 海参酸性黏多糖经酸水解产物色谱图Fig.3 Chromatogram of hydrolysis products of mucopolysaeccharide
2.4 线性范围
在本实验条件下,配制浓度为0~20 mg/L的系列海参多糖标准液,以吸光值为纵坐标,样品多糖浓度为横坐标,得到线性回归方程为:y=0.031x-0.0144,相关系数大于0.99,线性良好。
2.5 显色剂用量的确定
当样品中多糖含量过高(本试验中高于20 mg/L)时,亚甲基蓝用量不足,会导致测定结果偏低,反之,当亚甲基蓝用量过高(本实验中高于 1.0 mL)时,由于其具有显色干扰作用,也会导致测定结果偏低。通过改变亚甲基蓝用量测定酸性黏多糖含量的结果表明,当浓度为0.4 mmol/L的亚甲基蓝用量为1.0 mL时,该体系的吸光值变化最大,且稳定性良好,本实验选择亚甲基蓝用量为1.0 mL。
2.6 显色反应时间的确定
在分光光度法测定实验中,呈色反应的时间很有可能是影响测定结果的关键因素,本实验通过对浓缩冻干海参多糖样品的吸光值随时间变化情况进行测定,得到如图4结果。
从图4可以看出,该显色反应在50 min内吸光值的变化范围在 0.00~0.02之间,没有明显的下降趋势,这说明50 min内该显色反应稳定性持续良好;延长反应时间,溶液的吸光值呈现明显的下降趋势,此时溶液中逐渐析出颗粒状沉淀,且随着时间的延长,沉淀越来越多,溶液的吸光值明显降低,此时显色反应已经发生质的变化,不再适宜进行吸光值测定。考虑到显色反应的稳定性,本实验选择在30 min内对样品进行测试。
2.7 检测限
用分光光度法检测样品,以扣除空白值(未加标准的空白参比液吸光值为空白值)后的吸光值为0.01相对应的浓度值为检测限,即检出限L=(标准质量×0.01)/(标准吸光值-空白吸光值)[11]。采用此方法得到样品中海参酸性黏多糖的最低检出浓度为:0.79 mg/L。
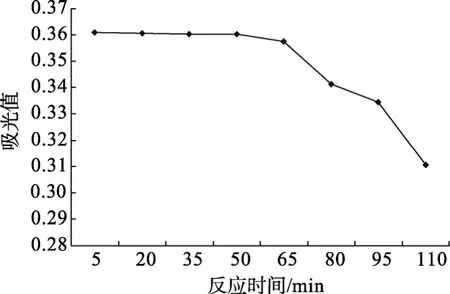
图4 海参多糖样品吸光值与反应时间关系Fig.4 Reaction kinetics
2.8 回收率试验
向已知多糖含量的样品中加入一定量多糖标准溶液,测定结果之差与加入多糖量之比即为回收率。
通过对测定方法的回收率考察发现,分别以海参三个组织样品为基底,添加不同水平的标准溶液时,其回收率在78.33%~100.09%范围内,能够满足测定要求(表1)。
2.9 不同组织多糖含量测定及方法精密度
通过表2的分析可以发现,海参体壁中酸性黏多糖含量最高,大约占总量的 4.751%,其次是海参卵,最少的是海参肠,这次实验结果也说明了酸性黏多糖不仅在海参体壁中含有,在其他部位也有相当可观的含量,因此在工业生产中可充分利用海参内脏中资源进行酸性黏多糖的提取加工。同时通过表2可以发现,通过对三个组织样品进行的5次重复测定,结果的相对标准偏差均小于 10%,这说明该方法的精密度符合要求。
3 结论
(1)本实验依据亚甲基蓝与海参酸性黏多糖反应能够产生异染配合物的特性,建立了海参酸性黏多糖含量测定的分光光度法,对显色剂用量以及反应时间等因素进行了探讨,确定了最佳的显色反应条件。实验中对该方法的检测限、回收率以及精密度都进行了测定,使该方法更科学完善。
(2)对海参中经济价值比较高的肠、卵以及体壁等部位进行了分别收集,并利用建立的方法对各部位中酸性黏多糖含量进行测定,最终发现海参体壁中多糖含量最高,海参卵次之,海参肠中多糖含量最少,这一测定结果,为海参酸性黏多糖工业化生产改进也提供了数据支持。
(3)针对不同水解方式对多糖含量测定结果的影响进行了探讨,最终发现采用木瓜蛋白酶水解的方法比采用酸水解方法更适合于分光光度法的测定。
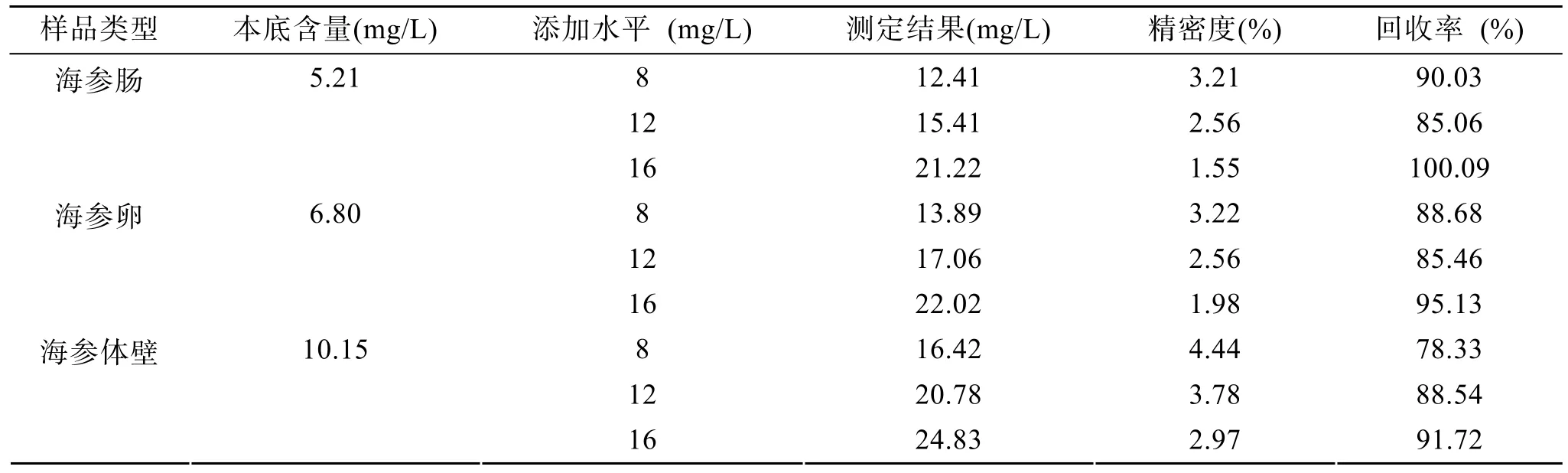
表1 测定方法的回收率Tab.1 Recoveries of spiking experiments
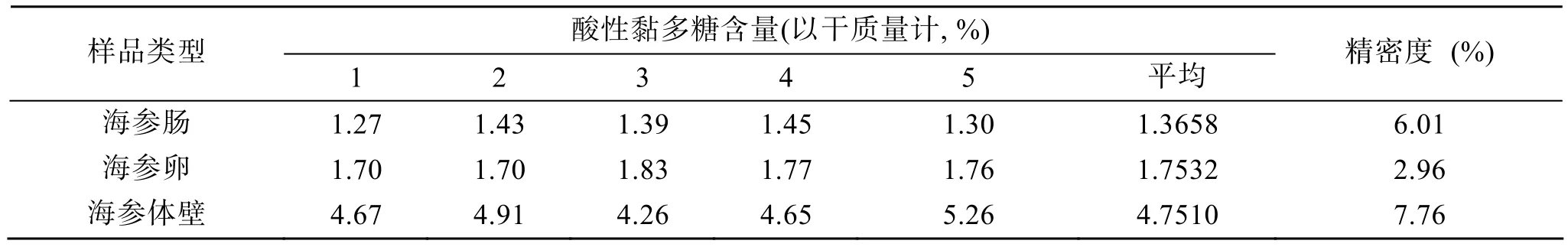
表2 样品测定及精密度Tab.2 Measurement and precision
[1]姜健,杨宝玲,邰阳.海参资源及其生物活性物质的研究[J].生物技术通讯,2004,5(15):537-540.
[2]王洪涛,付学军,申京宇.海参多肽、多糖综合提取工艺条件的优化[J].食品与生物技术学报,2006,25(6):83-86.
[3]Imanari T,Washio Y,Huary Y.Oral absorption and clearance of partially deploymerized fucosy chondroitinsulfate from sea cucumber[J].Thromb Res,1999,93(3):129-135.
[4]Nagase H,Enjyoji K,Minamiiguchi K.Deploymerized holothurian glycosminoglycan with novel anticoagulant actions:antithrombin Ⅲ and heparin cofactorⅡ-independent inhibitor of factor X activation by factor Ⅷ complex and heparin cofactor Ⅱ-dependent inhibition of thrombin[J].Blood,1995,85:1 527-1 534.
[5]李海棠,周清凯,蔡云见.薄层色谱扫描法测定海康胶囊中海参多糖含量[J].时珍国医国药,1999,8(10):579.
[6]李燕妮,车业娜.分光光度法测定海参多糖含量方法的改进[J].云南化工,2008,4(35):27-28.
[7]李桂芝,刘永明,秦华伟.亚甲基蓝褪色分光光度法测定藻酸双酯钠[J].分析化学研究简报,2005,9(33):1 324-1 326.
[8]刘红英.海带岩藻聚糖硫酸酯测定方法的研究[J].青岛海洋大学学报,2002,3(32):236-240.
[9]Soedjak H S.Colorimetric determination of carrageenans and other anionic hydrocolloids with methylene blue[J].Anal Chem,1994,66:4514-4518.
[10]郑艾初,陈建,彭超英.糙海参酸性黏多糖的提取纯化工艺探讨[J].现代食品科技,2007,23(5):65-67.
[11]王叔淳.食品卫生检验技术手册[M].3版.北京:化学工业出版社,2002.
猜你喜欢
意林彩版(2022年2期)2022-05-03
供水技术(2021年3期)2021-08-13
作文与考试·小学低年级版(2021年12期)2021-08-09
魅力中国(2020年8期)2020-12-07
金桥(2018年2期)2018-12-06
恋爱婚姻家庭(2018年33期)2018-07-22
现代园艺(2017年21期)2018-01-03
红蜻蜓·低年级(2014年9期)2015-03-23
物理化学学报(2015年5期)2015-02-28
云南中医学院学报(2014年5期)2014-07-31
