
氯霉素固体脂质纳米粒的制备及质量评价
2011-01-24 02:42聂绩黄华
中国药房 2011年17期
聂绩,黄华
(重庆医科大学药学院高校药物工程研究中心,重庆市 400016)
氯霉素固体脂质纳米粒的制备及质量评价
聂绩*,黄华#
(重庆医科大学药学院高校药物工程研究中心,重庆市 400016)
目的:制备氯霉素固体脂质纳米粒(CAP-SLN)并考察其质量。方法:选取CAP与甘油棕榈酸硬脂酸酯(Precirol ATO 5)比例(药脂比)、泊洛沙姆含量、乳化温度和初乳-分散相的体积比为考察因素,包封率和载药量为评价指标,设计正交试验并优化处方,利用乳化蒸发-低温固化法制备CAP-SLN;同时以粒径、Zeta电位、包封率、载药量、稳定性及体外释放度为指标评价其质量。结果:最佳制备处方药脂比为1∶10,泊洛沙姆含量为2%,乳化温度为70℃,初乳-分散相的体积比为1∶7。所制纳米粒平均粒径为227nm,Zeta电位为-30.5mV,平均包封率为65.9%,平均载药量为6.59%;于4℃环境中考察30d,其包封率、粒径无显著变化,25℃环境中包封率显著降低、粒径明显增大;在前4h内有明显突释现象,药物累积释放率达58.86%,48h时累积释放率达85.09%,体外释药行为符合Weibull方程。结论:该制剂处方设计和工艺方法可行,制剂质量符合要求,可达到缓释效果。
氯霉素;固体脂质纳米粒;甘油棕榈酸硬脂酸酯;体外释放度
固体脂质纳米粒(Solid lipid nanoparticles,SLN)是一种新型纳米粒给药系统[1],具缓/控释特性,可提高药物生物利用度及其稳定性等[2,3],适用于亲脂性药物的包裹[4]。氯霉素(Chloramphenicol,CAP)是治疗沙眼和结膜炎等眼部疾病最常用的抗生素之一,使用中为保证药效需每日给药3~5次,在全身用药时不良反应主要表现为骨髓抑制与再生障碍性贫血,其在生产和贮存中可产生二醇物,使抗菌活性降低。为此,本文选用甘油棕榈酸硬脂酸酯(Precirol ATO 5)为载体材料,以CAP为模型药制备CAP-SLN,并对其制备工艺、理化性质和体外释放度进行考察,以期达到缓释、减少给药次数、降低毒副反应、提高生物利用度的目的,为进一步研制CAP-SLN眼用混悬液提供参考。
1 仪器与试药
1.1 仪器
Zetasizer Nano ZS90激光粒度仪(英国马尔文仪器有限公司);Hitachi S-3400扫描电镜(日本日立公司);UV-3105型紫外可见分光光度计(日本岛津公司);DF-101S集热式恒温加热磁力搅拌器(巩义市予华仪器有限责任公司);82-2恒温磁力搅拌器(上海青浦沪西仪器厂);数显恒温水浴锅(涿州市国华电器有限责任公司);RCZ-6B1型药物溶出仪(上海黄海药检仪器厂)。
1.2 试药
CAP原料药(郑州二七长兴化工物资有限公司,批号:090625,含量:98.2%);CAP标准品(中国药品生物制品检定所,批号:130555-200602,含量:99.4%);Precirol ATO 5(法国Gattefosse公司);蛋黄卵磷脂(北京双旋微生物培养基制品厂);泊洛沙姆(F68,德国Basf公司);Sephadex-G50(瑞典Pharmacia公司);无水乙醇(重庆川东化工(集团)有限公司化学试剂厂);双蒸水、CAP-SLN(规格:0.05%)均由重庆医科大学药学院实验室制备。
2 方法与结果
2.1 CAP-SLN的制备
采用乳化蒸发-低温固化的方法制备。具体操作:精密称取处方量CAP原料药、Precirol ATO 5和蛋黄卵磷脂,置于25mL具塞锥形瓶中,加入10mL无水乙醇,超声约1min,使蛋黄卵磷脂充分溶解,置于70℃恒温水浴锅中保持恒温,构成有机相。精密称取处方量的泊洛沙姆,溶于适量的水中,使恒温保持在70℃,构成水相。用预热至约70℃的注射器缓慢地将有机相注入1300r·min-1搅拌速度下的恒温水相中,继续以此速度搅拌2~3h,形成半透明的初乳,迅速倒入0~2℃的一定量的分散相(与上述水相成分一致)中,继续搅拌2h,即得到半透明、显蓝色乳光的CAP-SLN溶液。
空白SLN的制备方法同上述CAP-SLN的制备方法,只是未加入CAP原料药。
2.2 含量测定
用无水乙醇制备浓度为15µg·mL-1的CAP原料药溶液,在200~400nm范围内进行紫外扫描,其最大吸收波长为273nm。精密称取一定量的空白SLN溶液,用无水乙醇破乳,并稀释至50mL,进行测定,发现在273nm波长处无紫外吸收,表明空白辅料对测定无干扰,方法专属性好。故采用紫外分光光度法测定含量,光谱图见图1。

图1 紫外吸收光谱图A.CAP原料药;B.空白SLNFig1 UV absorption spectrumA.CAP material;B.blank SLN
2.2.1 标准曲线的制备:精密称取CAP标准品5.0mg,置于50mL容量瓶中,用无水乙醇制备成浓度为0.1mg·mL-1的CAP标准品溶液,精密量取0.5、1.0、2.0、3.0、4.0和5.0mL标准品溶液,置于25mL容量瓶中,用无水乙醇稀释成浓度为2、4、8、12、16和20µg·mL-1系列溶液,以无水乙醇为空白,于273nm波长处测定吸收度(A)。以A对标准品溶液浓度(c)进行线性回归,得回归方程:A=0.0303c+0.0135(r=0.9999)。结果,CAP检测浓度的线性范围为2~20µg·mL-1。
2.2.2 回收率与精密度试验:用无水乙醇制备4、12、20µg·mL-13种浓度的CAP原料药溶液,各3份,加入相应处方量的空白SLN,测定A,计算回收率。结果平均回收率分别为100.4%、99.9%、99.8%,RSD分别为0.52%、0.27%、0.16%。选取同样的4、12、20µg·mL-13种浓度的CAP原料药溶液,各3份,测定A,计算日内RSD(同日内测5次)和日间RSD(连续5d测定)。日内RSD分别为1.00%、0.48%、0.30%,日间RSD分别为0.71%、0.26%、0.11%。回收率试验结果见表1。
2.3 包封率测定

表1 回收率试验结果(n=3)Tab1 Results of recovery tests(n=3)
采用葡聚糖凝胶柱分离-紫外分光光度法测定CAP-SLN的包封率。
精密量取1.0mL的CAP-SLN溶液,缓慢置于已装好的Sephadex-G50凝胶柱(1.5cm×30cm)上,经预试验考察后,选择流动相为水,流速为1.0mL·min-1,用试管收集流出液,每2mL收集1管,绘制分离曲线,详见图2。

图2 CAP-SLN和游离CAP分离曲线Fig2 Elution curves of CAP-SLN and free CAP
收集、合并有蓝色乳光溶液,置于25mL棕色容量瓶中,用无水乙醇破乳并稀释,以紫外分光光度法在273nm波长处测定A1。另精密量取1.0mL的CAP-SLN溶液,置于25mL棕色容量瓶中,用无水乙醇破乳并稀释,在273nm波长处测定吸光度A2。

2.4 正交试验
根据单因素试验结果,确定了主要影响因素:即药脂比(CAP与Precirol ATO 5的比例)(A)、泊洛沙姆含量(B,%)、乳化温度(C,℃)和初乳-分散相的体积比(D),以正交试验表L9(34)进行试验,以包封率和载药量为评价指标进行考察。正交试验因素水平见表2,正交设计试验结果见表3(由表2、表3可见,极差R反映各因素对指标影响的程度,R越大,影响程度越大)。

表2 因素水平表Tab2 Factors and levels
由表3结果分析,以包封率为衡量指标,影响程度排列为A>B>D>C,其中A3>A2>A1,B2>B3>B1,C2>C3>C1,D3>D1>D2,得出最佳优化处方A3B2C2D3。以载药量为衡量指标,影响程度排列为A>B>D>C,其中A1>A2>A3,B2>B3>B1,C2>C3>C1,D3>D2>D1,得出最佳优化处方 A1B2C2D3。从试验结果得出,当药脂比较大时(1∶15),药物包封率较高;药脂比较小时(1∶5),其载药量较高。经过综合考虑,确定A2为最佳药脂比,则最佳优化处方为A2B2C2D3,即药脂比为1∶10,泊洛沙姆含量为2%,乳化温度为70℃,初乳-分散相的体积比为1∶7。按最优处方制备3批样品,测得包封率分别为65.6%、66.3%和65.7%,平均包封率为65.9%,RSD=0.57%;载药量分别为6.56%、6.63%、6.57%,平均载药量为6.59%,RSD=0.57%。

表3 正交设计试验结果Tab3 Results of orthogonal design
2.5 形态观察
按最优处方制备的CAP-SLN溶液,于扫描电镜下观察,其形态为大小较均匀的类球形,详见图3。

图3 CAP-SLN扫描电镜照片(×30000)Fig3 Transmission electron microphotograph of CAP-SLN(×30000)
2.6 粒径和Zeta电位
用适量水稀释按最优处方制备的CAP-SLN溶液,用Zetasizer Nano ZS90激光粒度仪测定粒径和Zeta电位,测得平均粒径为227nm,Zeta电位为-30.5mV。粒径分布与Zeta电位结果详见图4、图5。
2.7 稳定性考察
重新制备3批样品(批号:100325-1、100325-2、100325-3),分别放入4℃和25℃的环境中,于0、5、15、30d观察其平均包封率和平均粒径的变化,考察其稳定性,结果见表4。

表4 稳定性试验结果Tab4 Results of stability tests
由表4可见,CAP-SLN在4℃储存温度下包封率和粒径无显著变化,稳定性良好;在25℃储存温度下包封率显著降低,粒径明显增大,稳定性较差。
2.8 体外释放度考察
精密量取4.0mL的CAP-SLN溶液,放入预处理好的透析袋中,两端用细线系紧,放入装有200mL释放介质的溶出杯中。根据眼内pH为7.4、眼表温度为34℃的常规,本试验采用以下条件:释放介质为pH 7.4磷酸盐缓冲液,水浴温度为34℃,转速为100r·min-1。将释放介质分别于0.5、1、2、4、8、12、24、48h取样5mL,每次取液后补加同体积的释放介质,用紫外分光光度法进行测定。采用相同方法考察CAP原料药的体外释放度,CAP原料药和CAP-SLN的累积释放曲线结果见图6。
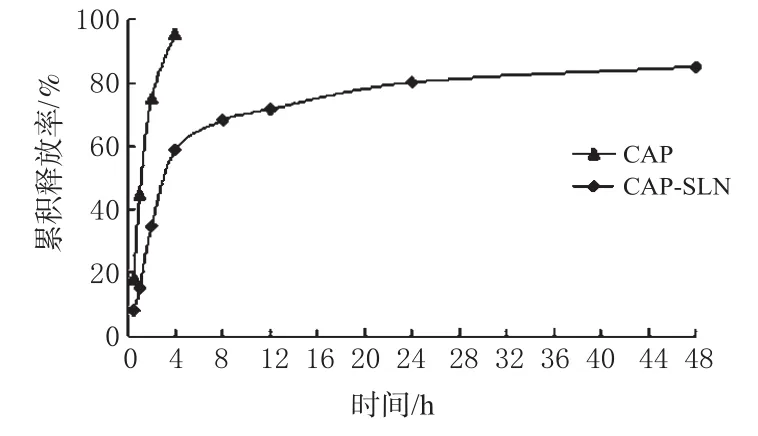
图6 CAP原料药和CAP-SLN累积释放曲线Fig6 Drug release curve of CAPand CAP-SLN
由图6可见,CAP原料药在4h内的累积释放率已达到95.6%;CAP-SLN在前4h内有明显的突释现象,药物累积释放率达到58.86%,而后缓慢释放药物,48h时累积释放率达85.09%。将CAP-SLN累积释放量(Q)和时间(t)进行拟合,结果符合Weibull方程:

3 讨论
3.1 CAP-SLN制备方法
本试验采用乳化蒸发-低温固化的方法制备样品,把药物、聚合材料和表面活性剂溶于半极性的且与水互溶的溶剂中,然后在磁力搅拌的基础上将溶液注入非溶剂(一般为含表面活性剂的水)中。由于溶剂的快速扩散,纳米粒随即形成。因CAP略溶于水,药物大部分存在于Precirol ATO 5所形成的乳滴中,在泊洛沙姆和蛋黄卵磷脂的乳化作用下形成CAP的纳米乳液。把乳液分散于0~2℃冰水中,乳液的温度急剧降到Precirol ATO 5熔点以下,乳液凝固成固态Precirol ATO 5纳米粒。如此可提高CAP的稳定性,避免产生化学降解。但也有人认为SLN是热力学不稳定系统,在较短的时间内就会产生粒径的增大、粒子聚合或胶凝[5]。Zeta电位也与纳米粒体系的稳定性密切相关,在-60~-30mV范围内体系物理性质稳定,因此测定Zeta电位可以预测纳米粒体系的储藏稳定性[6]。本试验制备的CAP-SLN的Zeta电位值为-30.5mV,属热力学稳定系统。该制剂处方设计和工艺方法可行,制剂质量符合要求,可达到缓释效果。
3.2 辅料和溶剂的选择
本试验采用的脂质是Precirol ATO 5。在考察脂质材料对CAP-SLN性质的影响时,选择了4种脂质材料:硬脂酸、单硬脂酸甘油酯、Compritol 888ATO和Precirol ATO 5。结果发现,硬脂酸作载体的CAP-SLN稳定性非常差,储存于4℃条件下,在4h内就开始浑浊,有絮状物产生;单硬脂酸甘油酯作载体的CAP-SLN稳定性好,但包封率低;Compritol 888ATO作载体的CAP-SLN包封率高,但稳定性低;相比而言,PrecirolATO 5作载体的稳定性和包封率均较好。Precirol ATO 5[7]其主要成分是硬脂酸和棕榈酸,毒性低于高分子合成材料,在体内可以降解且生理相容性好,同时理化性质稳定,可改变单一脂质晶格的高度有序性,使药物更易镶嵌于脂质晶格中,或是存在于脂质的脂肪酸链间、脂质层间,有利于药物的稳定。另外该材料熔点为60℃,相对较低,有利于CAP-SLN的制备和理化性质的稳定。
蛋黄卵磷脂是细胞膜组成成分,毒性低,可以在体内降解且生理相容性良好,当卵磷脂和泊洛沙姆合用时,泊洛沙姆可插入卵磷脂单分子层形成紧密充填的混合层,从而减小粒径,提高稳定性,故选其作为表面活性剂[8,9]。溶剂选择的是无水乙醇,满足半极性且与水互溶的性质。蛋黄卵磷脂也易溶于无水乙醇,有利于油相混合均匀。
3.3 体外释放度的测定
本试验采用透析法考察CAP-SLN的体外释放规律。眼用制剂的体外释放度考察应遵循眼表温度,所以选择34℃。从体外释放曲线可见,样品在初期有突释的现象,可能是由于存在未包入脂质材料的游离CAP,而产生的突释现象。药物的快速释放,可使CAP在眼部快速达到抑菌浓度,产生药效。由于大部分的药物包裹进入脂质材料的骨架结构中,从而缓慢地释放药物,所以8h后释放趋于平稳,达到了较好的缓释目的。后续试验将对CAP-SLN眼用制剂在兔眼内的药动学进行研究,考察其对兔眼的刺激性、在兔眼内的滞留时间和在兔眼泪液及房水内的药动学,对其在体内实现缓释、降低毒副反应、提高生物利用度的作用进行验证。
[1] Muller RH,Mader K,Gohla S.Solid lipid nanoparticles(SLN)for controlled drug delivery-a review of the state of the art[J].Eur J Pharm Biopharm,2000,50(1):161.
[2] Manjunath K,Reddy JS,Venkateswarlu V.Solid lipid nanoparticles as drug delivery systems[J].Methods Find Exp Clin Pharmacol,2005,27(2):127.
[3] Kalariya M,Padhi BK,Chougule M,et al.elobetasol propionate solid lipid nanopartides cream for effective treatment of eczema:formulation and clinical implications[J].Indian J Exp Biol,2005,43(3):233.
[4] Hu L,Tang X,Cui F.Solid lipid nanoparticles(SLNs)to improve oral bioabailability of poorly soluble drugs[J].J Kristl Pharm Pharmacol,2004,56(12):1525.
[5] Wang J,Chen DB,Zhang Q.Determination of surface properties and cell up take in vivo of stearic acid solid lipid nanoparticle[J].Chin Pharm J,2001,36(2):102.
[6] Chansiri G,Lyons RT,Patel M.Effect of surface charge on the stability of oil/water emulsions during steam sterilization[J].J Pharm Sci,1999,88(4):454.
[7] Wissing SA,Kayser O,Muller RH.Solid lipid nanoparticles for parenteral drug delivery[J].Adv Drug Delivey Rev,2004,56(9):1257.
[8] Heurtault B,Saulnier P,Pech B,et al.Physico-chemical stability of colloidal lipid particles[J].Biomaterials,2003,24(23):4283.
[9] Han F,Li SM,Yin R,et al.Effect of surfactants on the formation and characterization of a new type of colloidal drug delivery system:Nanostructured lipid carriers[J].Colloids and Surfaces A:Physieochemical and Engineering Aspects,2008,31(5):210.
Preparation and Quality Evaluation of Chloramphenicol Solid Lipid Nanoparticles
NIE Ji,HUANG Hua
(Chongqing Engineering Research Center for Medicine,College of Pharmacy,Chongqing Medical University,Chongqing 400016,China)
OBJECTIVE:To prepare Chloramphenicol solid lipid nanoparticles(CAP-SLN)and to study the quality of it.METHODS:The preparation technilogy of CAP-SLN was optimized by orthogonal test with the entrapment efficiency and drug loading amount as index and with proportion of CAP to Precirol ATO 5(drug-to-lipid ratio),amount of poloxamer,emulsifying temperature,volume ratio of emulsion-disperse phase as factors.CAP-SLN was prepared by emulsification evaporation-low temperature solidification technique.The quality of preparation was evaluated with particle size,Zeta potential,drug-loading amount,stability and in vitro drug release rate as index.RESULTS:The optimal formulation was as follows:drug-to-lipid ratio of 1∶10,the weight of poloxamer of 2%,emulsifying temperature of 70℃,drug-to-lipid of 1∶7.The mean diameter of CAP-SLN was 227nm.The zeta potential was-30.5mV.The entrapment efficacy was 65.9%.The average drug-loading amount was 6.59%.CAP-SLN could keep stable at 4℃at least for one month.The entrapment efficacy of CAP-SLN at 25℃decreased significantly while particle size increased.The burst release of CAP-SLN was found during the former 4h.The drug release rate of it was 58.86%at 4h and reached 85.09%at 48h.The in vitro drug release behavior was in line with Weibull equation.CONCLUSION:The preparation technique and formulation are practicable.CAP-SLN is up to quality standard and can achieve sustained-release effects.
Chloramphenicol;Solid lipid nanoparticles;Precirol ATO 5;In vitro drug release rate
R927.11;R944.1+6
A
1001-0408(2011)17-1598-04
*硕士研究生。研究方向:速释及缓、控释制剂。电话:023-68485078
#通讯作者:教授,硕士研究生导师。研究方向:速释及缓、控释制剂。电话:023-68485078
2010-08-03
2010-09-20)
·药品监督·
猜你喜欢
发明与创新(2022年31期)2022-11-03
中国医药科学(2022年5期)2022-05-05
中国盐业(2018年20期)2019-01-14
中成药(2018年9期)2018-10-09
中成药(2018年1期)2018-02-02
中成药(2017年4期)2017-05-17
中国继续医学教育(2015年6期)2016-01-07
中国当代医药(2015年33期)2015-03-01
中华皮肤科杂志(2014年4期)2014-12-19
中华皮肤科杂志(2014年3期)2014-12-19
