
乌药产地加工的研究
2010-08-28 03:32杨立平邓桂明欧阳荣
中国现代药物应用 2010年14期
杨立平 邓桂明 欧阳荣
乌药为樟科植物乌药 Lindera strychnifolia(Sieb.ef Zucc.)Vill.的干燥块根[1],乌药中主要含有挥发油、异喹啉类生物碱及呋喃倍半萜三类主要成分[2],共同构成了乌药的化学特征。乌药为历代中医临床常用的理气药,功能理气止痛、温肾散寒[3]。
乌药的现代研究多注重于生药的化学成分及药理作用的研究,对于产地加工、干燥工艺对成分的影响,尚无系统研究报道。本实验结合现代技术手段,对乌药的产地加工工艺进行研究,通过比较不同温度、摊层厚度、干燥时间等与干燥工艺直接相关的因素,采用乌药主要有效成份乌药醚内酯为指标[4],得出了乌药产地加工工艺的规范化工艺参数。该研究方法新颖,手段先进,为乌药炮制的规范化研究提供参考。
1 仪器与试药
Agilent110高效液相色谱仪(安捷伦科技有限公司);电热恒温鼓风干燥箱(上海跃进医疗器械厂);不锈钢电热恒温水浴锅(北京市医疗设备厂);KQ-600型超声波清洗器(昆山市淀山湖检测仪器厂);植物粉碎机(泰特仪器有限公司);水分测定装置。
甲醇、乙腈为色谱纯(天津市密欧化学试剂有限公司);甲苯、无水乙醇、香草醛、石油醚、乙酸乙酯、硫酸等为分析纯;水(灭菌用水,自制)。
乌药药材,采于湖南衡东,经湖南中医药大学第一附属医院药检室鉴定为樟科植物乌药Lindera strychnifolia(Sieb.ef Zucc.)Vill.的干燥块根。
2 实验方法与结果
2.1 产地新鲜药材切制工艺的考察
2.1.1 乌药饮片含水量曲线的制备
2.1.1.1 样品的制备 药材净取后直接切片,以3 kg/m2(约1 cm厚)、6 kg/m2(约2 cm厚)、9 kg/m2(约3 cm厚)的厚度摊层,分别在60℃、80℃、100℃温度下,烘箱中干燥,每1 h取样一次。从所取样品中称取10 g,粗粉碎后按2005年版《中国药典》附录中之甲苯法测饮片含水量。(见表1)。

表1 不同温度及摊层厚度下乌药含水量测定
2.1.1.2 结果分析 结果表明:乌药在常压下80℃、100℃时3 kg/m2、6 kg/m2组在3 h后含水量均趋于稳定,而9 kg/m2组趋于恒定的时间各不相同。
2.1.2 乌药醚内酯的含量测定
2.1.2.1 色谱条件 流动相:梯度洗脱(甲醇:乙腈:水)梯度洗脱,洗脱梯度见表2;波长235 nm;柱温:35℃;流速:1 ml/min。

表2 梯度洗脱表
2.1.2 对照品的制备 精密称定乌药醚内酯对照品2 mg,置25 ml容量瓶中,用甲醇溶解并稀释至刻度,摇匀,即得。(每1 ml中含乌药醚内酯80 μg)。
2.1.3 样品制备
2.1.3.1 浸泡时间的考察 乌药粉碎成细粉,取1 g,每个供试品取8份,分别精密称定,置250 ml锥形瓶中,参照《中国药典》2005年版一部乌药含量测定项下所用溶剂,各精密加入甲醇 50 ml,称重重量,分别浸泡 0 h、1 h、4 h、24 h 后,超声15 min,放冷,再称定重量,并用甲醇补足减失的重量,摇匀,用0.45 μm微孔滤膜过滤,弃去初滤液,取续滤液,备用。结果表明乌药浸泡0 h和1 h时乌药醚内酯的含量比较高,但两者的含量相差不大,而4 h、24 h含量降低,所以实验过程中选择将药材粉末浸泡0 h后进行提取。
2.1.3.2 超声时间的考察 乌药粉碎成细粉,取1 g,每个供试品取10份,分别精密称定,置250 ml锥形瓶中,参照《中国药典》2005年版一部乌药含量测定项下所用溶剂,各精密加入甲醇50 ml,称重重量,分别振摇10 min、超声15 min、30 min、60 min、90 min,放冷,再称定重量,用甲醇补足减失的重量,摇匀,用0.45 μm微孔滤膜过滤,弃去初滤液,取续滤液,备用。结果表明,超声提取比手动振摇的效果好,且超声15 min效果最好。
2.1.3.3 提取方法的考察 乌药粉碎成细粉,取1 g,每个供试品取4份,分别精密称定,置250 ml锥形瓶中,参照《中国药典》2005年版一部乌药含量测定项下所用溶剂,加甲醇50 ml,称重,一份超声 15 min,放冷,再称定重量,用 0.45 μm微孔滤膜过滤,取续滤液备用。另一份用索氏提取器在60℃提取4 h,所得固体用甲醇定容至10 ml容量瓶中,用0.45 μm微孔滤膜过滤,取续滤液备用。结果表明,用超声提取15 min比用索氏提取器提取效果更好。
2.1.3.4 提取次数的考察 乌药粉碎成细粉,取1 g,每个供试品取4份,分别精密称定,置250 ml锥形瓶中,参照《中国药典》2005年版一部乌药含量测定项下所用溶剂,加甲醇50 ml,称重,第1份超声提取一次,超声15 min,再称定重量,用甲醇补足减失的重量,用0.45 μm微孔滤膜过滤,弃去初滤液,取续滤液备用。第2份超声提取二次,分别15 min,第3超声提取三次,其他操作同上。结果表明,超声提取次数对乌药醚内酯的含量影响不大,为节约时间,故超声提取一次。
2.1.3.5 样品提取方法 取本品细粉约1 g,精密称定,置250 ml锥形瓶中,超声提取二次,每次加甲醇25 ml,超声15 min,合并滤液至50 ml量瓶中,并加甲醇至刻度,摇匀,用0.45 μm微孔滤膜过滤,弃去初滤液,取续滤液,备用。
2.1.4 标准曲线的制备 分别精密吸取各浓度的乌药醚内酯对照品溶液10 μl,其浓度分别为 0.016 mg/ml、0.032 mg/ml、0.048 mg/ml、0.060 mg/ml、0.080 mg/ml、0.160 mg/ml注入高效液相色谱仪,记录色谱图,测定峰面积,以进样量为横坐标,峰面积值Y为纵坐标绘制标准曲线,并计算回归方程:Y=3469.84575X-2.73808,r=0.9997。结果表明,乌药醚内酯在0.16 μg/ml~1.6 μg范围内峰有较好的线性关系。
2.1.5 精密度实验 分别精密吸取对照品溶液10 μl,重复进样6次。其乌药醚内酯峰面积的RSD为1.34%。
2.1.6 稳定性试验 取当日配置的供试品溶液,于0 h、1 h、2 h、4 h、8 h、24 h 分别进样 10 μl,测定样品中乌药醚内酯峰面积的RSD值为3.36%。试验结果表明,供试液在8 h内稳定性良好。
2.1.7 重复性试验 取同一批供试品,按3.1.3项下方法,分别制备供试液6份,每份进样10 μl,测定峰面积并计算含量,结果RSD=1.31%。
2.1.8 加样回收率 称取已知含量的供试品(含12.00%的水分,含量1.3718 mg/g)约0.33 g6份,精密称定,分别置50 ml容量瓶中,分别精密加入0.1000 mg/ml乌药醚内酯对照品溶液5 ml,用甲醇稀释至刻度。按2.1.3项下操作,分别制得供试液,以上述色谱条件测定乌药醚内酯的含量,乌药醚内酯的平均回收率为103.7%,RSD为1.49%。(见表3)。
2.1.9 样品的测定 取供试品,按2.1.3项下的操作,进行测定,结果见表4。

表3 加样回收率试验(n=6)
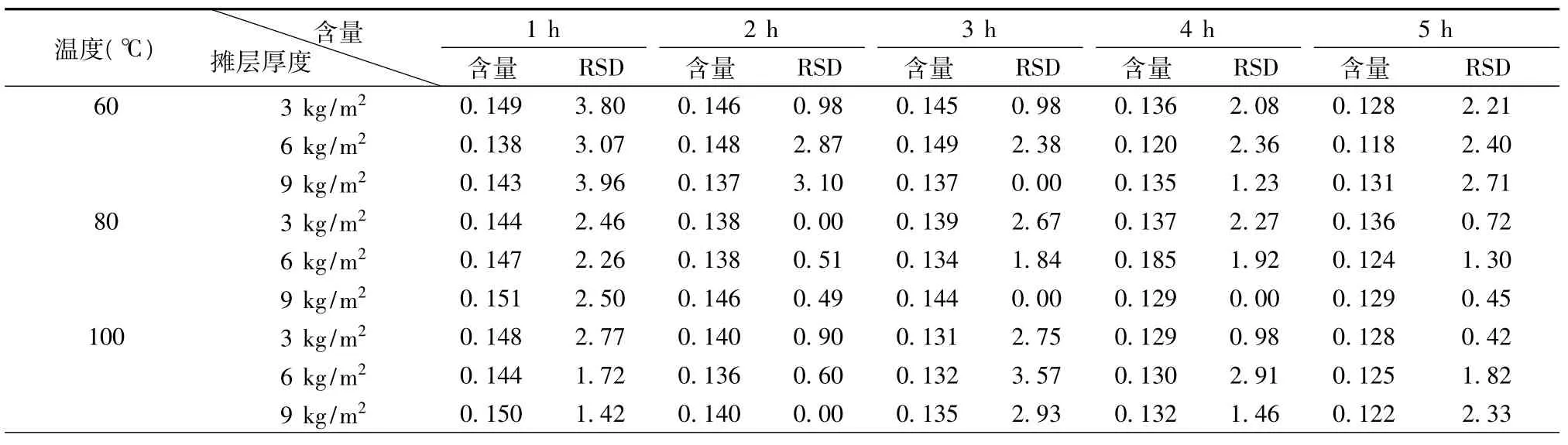
表4 不同温度及摊层厚度下乌药醚内酯的测定(g,%)
2.2 干燥药材切制工艺的考察
2.2.1 浸润方法的考察 将乌药干燥药材取四份,分别进行减压润、减压泡、常压润、常压泡的过程后进行切片,按上述样品制备方法制备供试品溶液,测得乌药醚内酯含量分别为 0.128 mg/g、0.148 mg/g、0.176 mg/g 和 0.148 mg/g。
2.2.2 结果分析 干燥后的乌药在常压下润湿后切片的加工方法所测得的乌药醚内酯含量比常压浸泡和减压条件下润湿,浸泡后切片的加工方法所测得的乌药醚内酯含量高。
3 讨论
3.1 干燥对含挥发性成分的药材的质量影响比较大。而现代对中药材干燥工艺的研究很少。本实验通过测定不同干燥温度、干燥时间下乌药醚内酯的含量,结果表明,温度对乌药醚内酯的含量影响较大。
3.2 通过上述试验结果表明乌药因其产地加工不同有效成分存在差异,采收后的乌药直接加工所含有的乌药醚内酯含量比较高。
3.3 实验过程中对湖南乌药细根部位和根部的非药用部位分别进行了考察,发现乌药除根茎部位外细根与其根部的非药用部位所测得的乌药醚内酯含量也符合药典规定。
3.4 本实验在进行浸泡时间考察时,乌药浸泡0 h和1 h时乌药醚内酯的含量比较高,但两者的含量相差不大,所以实验过程中选择将药材粉末浸泡0 h后进行提取。
[1]国家药典委员会.中华人民共和国药典.化学工业出版社,2005:58.
[2]杜志谦,夏华玲,江海肖.乌药挥发油化学成分的GC-MS分析.中草药,2003,34(4):308.
[3]王军伟,阮冰.乌药的植化及药理研究概况.浙江中医杂志,2006,11(41):675.
[4]程显隆,魏锋,冯玉飞,等.RP-HPLC法测定乌药中乌药醚内酯和乌药内酯的含量.药物分析杂志,2003,23(3):225.
猜你喜欢
科学家(2021年24期)2021-04-25
生物学通报(2021年4期)2021-03-16
陶瓷学报(2020年3期)2020-10-27
发明与创新·小学生(2019年4期)2019-04-19
中国环境监测(2019年1期)2019-03-13
中成药(2018年7期)2018-08-04
中国现代教育装备(2018年10期)2018-07-02
中成药(2017年10期)2017-11-16
中国卫生标准管理(2015年2期)2016-01-14
浙江中西医结合杂志(2015年12期)2015-06-06
