
疑似肝豆状核变性ATP7B基因第8外显子的基因序列分析1例
2010-08-21 00:20金银实南光贤
中国实验诊断学 2010年1期
金银实,胡 毅,王 佳,南光贤*
(1.吉林大学中日联谊医院神经内二科,吉林长春 130033;2.吉林大学畜牧兽医医学院,吉林长春 130062)
疑似肝豆状核变性ATP7B基因第8外显子的基因序列分析1例
金银实1,胡 毅1,王 佳2,南光贤1*
(1.吉林大学中日联谊医院神经内二科,吉林长春 130033;2.吉林大学畜牧兽医医学院,吉林长春 130062)
*通讯作者
肝豆状核变性(hepatolenticular degeneration,HLD)又称wilson病(Wilson‘s disease,WD),是由铜代谢障碍引起的常染色体隐性遗传性疾病。肝豆状核变性的致病基因为ATP7B基因,该基因的缺陷将会影响所编码的ATP7B活性,使过量的铜沉积在肝、脑、肾和角膜等组织器官,导致急慢性肝病、神经系统疾病、肾病和角膜色素环等。我国学者研究表明,8号外显子的Arg778Leu/Glu突变为我国ATP7B基因突变的第一热区,40.9%的肝豆状核变性患者可以检测到该突变,染色体突变率为30.7%[1]。血清铜蓝蛋白水平、K-F环、24小时尿铜含量、肝铜含量等是诊断WD的常用指标,简单凭借任何一项或几项指标不能确定或排除WD。其临床症状、体征复杂多样,往往造成诊断的困难[2-4]。肝豆状核变性也是可以有效治疗的遗传病之一,其疗效及预后与开始治疗时间密切相关[5]。如果能在发病早期甚至发病前确诊,通过药物治疗可以控制病情发展,提高患者的生活质量。反之,如若未能及时确诊,往往会延误最佳治疗时间,导致患者肝、脑、肾等组织器官发生不可逆性损害,致残甚至死亡。本文通过对1例疑似肝豆状核变性病例行遗传基因分析的方法来确诊,并指导治疗。
1 资料与方法
1.1 病例资料
杨XX,男,20岁。因右上肢颤动进行性加重2年余于2009年2月20日入我院。该患缘于2年前无明显诱因出现右上肢震颤,呈进行性加重,静止时正常,持物或活动时震颤明显,愈紧张愈明显,震颤幅度成进行性加重,现走路尚可,持物费劲。病程中伴有双下肢色素沉着。为求明确诊治遂入我院。病程中偶有饮水返呛,饮食及睡眠良好,大小便如常,体重减轻。既往健康。否认遗传病家族史,三代以内无类似发病患者。入院时查体:血压120/80 mmHg,面色晦暗,肝脏下缘达到肋弓一下3cm左右,未触及脾,双下肢黑色色素沉着,神清语明,示齿时双侧面纹对称,伸舌居中,四肢肌张力、肌力正常,四肢腱反射活跃,双侧有踝阵挛,双侧Babinski征阳性。血清铜蓝蛋白正常。头部CT未见异常。裂隙灯下角膜未发现K-F环。
1.2 研究对象
病例组为疑似肝豆状核变性患者及其母亲血样,加入肝素抗凝备用。正常组为无肝豆状核变性疾患的正常人。
1.3 基因检测
1.3.1 制备基因组DNA取外周血5 ml,加入肝素抗凝。
1.3.2 全基因组的提取 使用由V-gene公司购置的Whole Blood Genomic DNA Mini-prep Kit血液基因组提取试剂盒按说明书要求提取全基因组。提取的DNA样品用0.8%琼脂糖凝胶电泳检测提取效果。
1.3.3 人ATP7B基因序列第8外显子序列的获得从 GenBank(http://www.ncbi.nlm.nih.gov/)检索人ATP7B基因序列及其第8外显子核酸序列。第八外显子全部序列如下:tcctctgaatgggaaagtatatttcataaacgcccatcacagaggaagaagtactgtcacgactgtgcacaaagctagaggctttgccatccccagggcccttggccctgtgtcgctcattgaactctcctccctacttgctggcagccttcactgtccttgtctttcagctcctcggtgggtggtacttctacgttcaggcctacaaatctctgagacacaggtcagccaacatggacgtgctcatcgtcctggccacaagcattgcttatgtttattctctggtcatcctggtggttgctgtggctgagaaggcggagaggagccctgtgacattcttcgacacgccccccatgctctttgtgttcattgccctgggccggtggctggaacacttggcaaaggtaacagcagcttcaggttcagaaaagagctgctccttcagtaaacaaatctcacttcctctgaacaccatgttta 截取ATP7B全基因序列57 781-58 260 bp,exon8编码区为57 951-58 184 bp(黑色斜体标出),长度为234 bp。
1.3.4 设计扩增第8外显子片段引物序列 利用Premier 5.0程序设计克隆引物序列,上游引物序列为5'-tat ttc ata aac gcc cat cac-3',下游引物序列为5'-gga agt gag att tgt tta ctg aag-3'。根据设计的引物推测,PCR产物理论长度约为450 bp。
1.3.5 从全基因组中PCR扩增ATP7B exon8目的基因 PCR扩增全部由Gene Amp PCR System 9600完成,启动子区的PCR扩增反应体积为50 μ L,反应液含:基因组DNA2 μ L,4 种 dNTP 混合液 4 μ L,DNA聚合酶 1 μ L,10 ×Ex Taq buffer5 μ L,4ddH2O 34μ L 。PCR扩增条件为94℃变性5 min,然后以下列温度和时间做35个循环:94℃30秒、62℃30秒、72℃30秒,最后72℃延伸7分。
1.3.6 PCR产物检测 PCR产物用1.0%琼脂糖凝胶电泳检测扩增片段大小是否符合要求。
1.3.7 PCR产物原液测序 将PCR产物原液交由上海生工生物工程有限公司进行测序。
1.3.8 测序结果比对 应用DNAMAN Version 5.2进行核酸及蛋白质序列比对。
2 结果
2.1 WD血液全基因组的获得 使用Whole BloodGenomic DNAMini-prep Kit血液基因组提取试剂盒提取人血液全基因组。0.8%琼脂糖凝胶电泳检测(图1)。
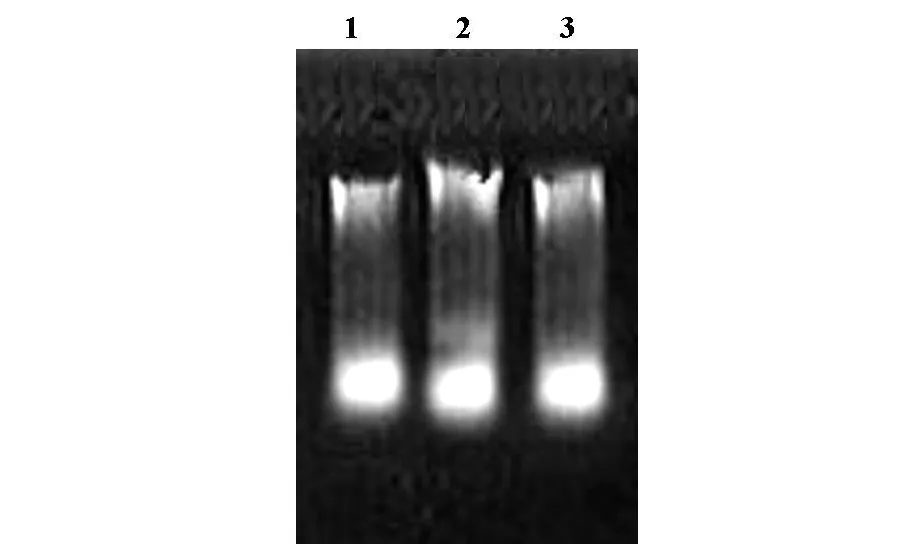
图1 人血液全基因组提取电泳分析
2.2 WD基因exon8基因片段的获得 以人血基因组为模板,Forward primer和Reverse primer为引物进行PCR扩增人ATP7B基因exon8片段,PCR产物理论长度约为450bp可见条带。1.0%琼脂糖凝胶电泳检测(图2)。
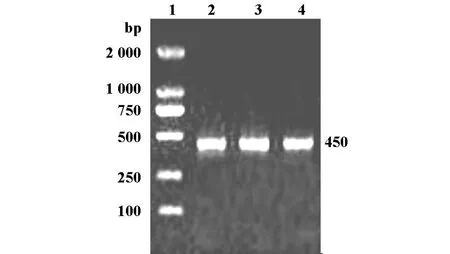
图2 人ATP7B基因exon8片段PCR扩增电泳分析
2.3 ATP7B基因exon8基因片段测序结果 通过PCR产物测序检测出的突变样品,经核算比对证实患者ATP7B基因exon8存在Arg778Leu/G2333T错义突变,同时伴随Leu700Leu/C2310G同义突变。
3 讨论
肝豆状核变性是较为常见的常染色体隐性遗传铜代谢障碍疾病,该病通常发生于儿童期和青少年期,发病年龄为5-40岁之间,但20岁以前发病者较多,男女无差别,典型病例头部CT及MRI双侧豆状核区可见对称性影像改变,血清铜蓝蛋白显著降低和尿铜排出量增高[6]。本文中疑似病例为青年男性,慢性起病,既往健康,否认遗传病家族史,主要表现为锥体外系症状,无肝脏症状,但触诊时发现肝大,无脾大,腹部彩超证实肝大,未发现脾大。裂隙灯下查眼部未发现K-F环。从发病形式及临床表现来分析高度怀疑肝豆状核变性,但缺乏诊断依据。肝豆状核变性为为数不多的治疗有效的遗传性疾病,若能获得早期诊断,恰当的长期系统驱铜治疗,就可使患者享受与健康人一样的生活和寿命。基因诊断被认为是早期确诊WD的最佳方法。该病的致病基因为ATP7B基因,ATP7B已被定位于13q14.3区。近年来的研究证实中国患者的突变热区为ATP7B8号外显子的778密码子[7,8]。笔者通过对疑似肝豆状核变性患者及其母亲、健康人的ATP7B外显子进行基因测序的方法来证实此患者存在8号外显子的基因突变,而此突变可能为导致发病的原因。在本文中,对病例组及对照组的ATP7B基因exon8进行PCR扩增产物测序结果显示:对照组的ATP7B基因片段序列与从GenBank检索到的序列完全一致;疑似肝豆状核变性患者的第8外显子存在纯合Arg778Leu/G2333T错义突变,同时伴随Leu700Leu/C2310G同义突变;患者母亲的ATP7B基因第8外显子存在杂合的Arg778Leu/G2333T错义突变,并伴随Leu700Leu/C2310G同义突变。
根据获得的ATP7B基因第8外显子测序结果,推测其基因表达产物进行氨基酸比对结果显示,可能存在Arg778Leu突变。而此突变可能为此患者发病的原因。
Arg778Leu位于ATP7B基因第8外显子,该区编码P型铜转运ATP酶的第四跨膜功能区(即Tm4),此区域只要负责铜离子的转运,Arg778Leu突变可能破坏该蛋白的一级、二级结构,影响Tm4的形成,使其功能丧失,若其他六个跨膜功能区仍具有功能,则其突变后果只能是削弱了铜的转运。
综上笔者对疑似肝豆状核变性患者exon8进行检测的方法来指导临床诊断,进行早期干预。基因序列检测对于基因快速筛选及早期快速确诊具有重大意义。若能够在发病早期或对高危人群进行诊断,降低患病率及死亡率。若能在产前进行诊断,则可以降低患儿的出生率,这对提高出生人口素质有极大推动作用。
[1]陈 源,耿江辉,张会丰.肝豆状核变性分子生物学研究进展.临床荟萃,2006,21(2):149.
[2]曹海燕,朱崇尧.肝豆状核变性的首发症状与误诊[J].罕少疾病杂志,2002,9(5):24.
[3]赵启华,贾云义.肝豆状核变性随访 15年1例[J].罕少疾病杂志,2002,9(6):51.
[4]张国范,侯太辉,王 龙.33例青少年肝豆状核变性误诊分析[J].罕少疾病杂志,2001,8(3):28.
[5]Yaize JG,Maitin P,Minox SJ,et al.Wilson discase:current status[J].Am J Med,2002,92:643.
[6]吴 江.神经病学.北京:人民卫生出版社,2005,257.
[7]吴志英,王 柠,林珉婷,等.Wilson病基因全长外显子的突变检测利分析.中华神经科杂志,2001,34:152-153.
[8]赵 鹏,张本恕.肝豆状核变性ATP7B基因Arg778Leu突变型与临床表现的相关性研究.脑与神经疾病杂志,2007,15(2):104.
1007-4287(2010)01-0110-03
2009-03-08)
猜你喜欢
中国造纸(2022年9期)2022-11-25
中国造纸(2022年8期)2022-11-24
电子科技大学学报(2022年5期)2022-10-29
现代临床医学(2021年4期)2021-07-31
中国生殖健康(2020年4期)2021-01-18
环球时报(2019-04-03)2019-04-03
中兽医学杂志(2019年1期)2019-01-06
中国生殖健康(2018年4期)2018-11-06
家庭医学(2017年4期)2017-05-27
湖北农业科学(2014年11期)2014-09-10
