
核酸适体电化学传感器研究的新进展
2010-06-26 06:03黄田贞林馨馨陈媛媛宦双燕
化学传感器 2010年1期
黄田贞,林馨馨,陈媛媛,宦双燕*
(1.湖南博云东方粉末冶金有限公司,湖南长沙410295)
(2.湖南大学化学化工学院,化学生物传感与计量学国家重点实验室,湖南长沙410082)
0 引言
核酸适体是一种能够与蛋白质、小分子、离子、核酸、甚至整个细胞等目标分子结合的人工合成寡聚核苷酸或肽分子。自从90年代Joyce,Gold和Szostak三个独立的研究小组做出了开创性工作之后[1~3],核酸适体的研究工作得到了快速发展。经过体外选择和扩增技术,从随机寡核酸文库中筛选出能与各种配体特异性结合的寡聚核苷酸片段,Gold小组把这种组合化学选择技术称为指数富集配体系统进化技术(Systematic Evolution of Ligands by Exponential Enrichment,SELEX)[3]。关于指数富集配体系统进化技术的详细进程已经有许多报道,在此不做详细介绍。
尽管关于核酸适体的研究目前还处于起始阶段,但作为生物传感器设计的传感元件方面,与抗体相比,核酸适体具有一定的优势[4~6]。第一,高亲和力。据报道,一些核酸适体与目标蛋白的亲和力(解离常数在皮摩到纳摩之间),可与单克隆抗体相媲美或比其更好。可通过提高筛选技术来提高亲和力、稳定性和结合特异性。一旦识别出寡核苷酸序列就可合成高纯度的核酸适体。第二,由于核酸适体的合成没有涉及到动物宿主,所以那些没有引发最小触发免疫反应的分子可以用来合成高亲和力的核酸适体。因此核酸适体的一个显著特征就是目标分子范围广。不仅寡核酸、蛋白质、酶和抗体可以用来制备核酸适体,一些辅基(磷酸腺苷,黄素单核苷酸等)、有机小分子(染料,药物,生长因子,氨基酸,肽,糖类等)、甚至是整个细胞、孢子和毒素也可用于核酸适体的合成。如果寡核苷酸库足够大,通过SELEX技术理论上可以筛选出针对任何目标物的核酸适体。第三,可以对核酸适体链在精确位点上进行官能团的修饰达到固定化的目的,而不影响其亲和力。另外,具有高稳定性。核酸适体可以进行不破坏其结构的可逆变性,而且在长期贮存和常温运输过程中也稳定。与抗体不同,核酸适体能够耐受非生理状态的pH值、温度和离子强度。此外,由于其小尺寸和低空间位阻,核酸适体在高密度微阵列传感器方面的应用优于抗体。
核酸适体可形成一些稳定的三维空间结构,如发夹、假结、凸环、G-四联体等结构。分子识别时,在目标分析物诱导下单链核酸适体会折叠成特殊的二级结构。分子识别的主要相互作用力是氢键、疏水作用和范德华力等[7~8]。这种适配识别称为适应性键合作用。核酸适体的出现为生物分析方法和传感器的设计开辟了新的思路。研究者们已经成功地创建了大量基于核酸适体的分析新方法,包括比色法,压电,表面等离子体共振(SPR),荧光,电化学原理等。该文重点介绍基于核酸适体电化学传感器的最新研究进展。
电化学传感器是应用最广泛和最古老的传感器类型之一,具有成本低、快速检测、灵敏度高、便携、操作方便、灵活、能独立运行、能自动和实时监测等显著优点。将核酸适体识别元件与各种电化学转换器相结合构建的新一代核酸适体传感器,集成了核酸适体和电化学传感器两方面的优势,成为近年来的一个研究热点。各种新颖的设计思路不断涌现,使电化学核酸适体传感器的性能不断得到改善,来解决分析应用中所面临的一些问题。下面就响应原理和存在的问题讨论如何设计一个性能优良的电化学核酸适体传感器。
1 亲和型核酸适体生物传感器
核酸适体是化学合成的寡核苷酸,可以很容易在核酸适体的精确位置完成一些功能团(-SH,-NH2,-COOH等)的修饰,从而便于在检测表面固定理想的核酸适体。通过自组装或共价连接,在不失去亲和力的情况下有序地完成核酸适体的固定化。亲和型生物传感器的基本策略是利用核酸适体和目标分子间的特殊亲和力直接设计。这种电化学核酸适体传感器可以分为两类:非标记型和标记型,如图1所示。
1.1 非标记型电化学核酸适体传感器
1.1.1 阻抗型(EIS)电化学核酸适体传感器
阻抗型电化学核酸适体传感器是一种无需加入额外的试剂或标记物的新型电化学生物传感器。制备EIS核酸适体传感器,核酸适体的固定化程序至关重要。一种方法是制备巯基自组装形成的混合单分子膜,即巯基短链分子和巯基核酸适体的杂化膜。混合膜可以减少电子转移电阻,并为目标物的结合提供空间。另一种方法是在紧凑的自组装单层膜上共价固定核酸适体。这样,由于目标分析物与电极表面间非特异性吸附的消除可以获得稳定的低噪声背景干扰。
在核酸适体传感器系统中,如果电子转移阻抗的变化被认为是关键的影响参数,可以采用[Fe(CN)6]3-/4-探针来监测这一识别过程,也称法拉第阻抗谱。通过监测电子转移电阻的变化可以直接灵敏的观察固定核酸适体与目标分析物之间的识别过程。被捕获的目标分析物能阻挡或促进电子转移。
Wang的小组报道了第一例识别诱导表面电荷转换的FIS适体传感器[9]。研究结果表明,如果体系 pH(pH 值=7.0)低于目标蛋白 pI(溶菌酶,pI=11),则蛋白质的识别将扭转表面电荷状态,促进电子转移,因此会降低电子转移电阻。然而,在大多数关于FIS核酸适体传感器的报道中,蛋白质识别是会阻碍电子转移的。徐等是第一例在短链阻塞模式下进行FIS核酸适体传感器研究[10]。混合杂化核酸适体膜表面结合IgE会增加阻抗。在该研究中还比较了基于抗体的生物传感器,结果表明,由于核酸适体体积小,结构简单,在减少背景噪声和获得更高的信号方面,基于核酸适体的生物传感器更有优势。他们采用该方法构建的IgE适体传感器,检测限为0.1 nmol/L。此后,Radi等做了相似研究[11],他们发现用2.0 mol/L NaCl处理10 min后,核酸适体感应表面可再生,并且经过至少15次重复检测再生后,电极行为并没有很大变化。Hsing等[12]利用FIS建造了用于检测凝血酶的核酸适体传感器。他们报道了4个数量级的检测范围,检测限为0.1 nmol/L。为了提高FIS核酸适体传感器的灵敏度,Fang的小组发现了一个放大电阻信号的方法[13]。目标蛋白结合后,利用盐酸胍使捕获的凝血酶变性导致凝血酶的水合半径增加,从而增大了界面电子转移电阻,检测限达到1.0×10-14mol/L。
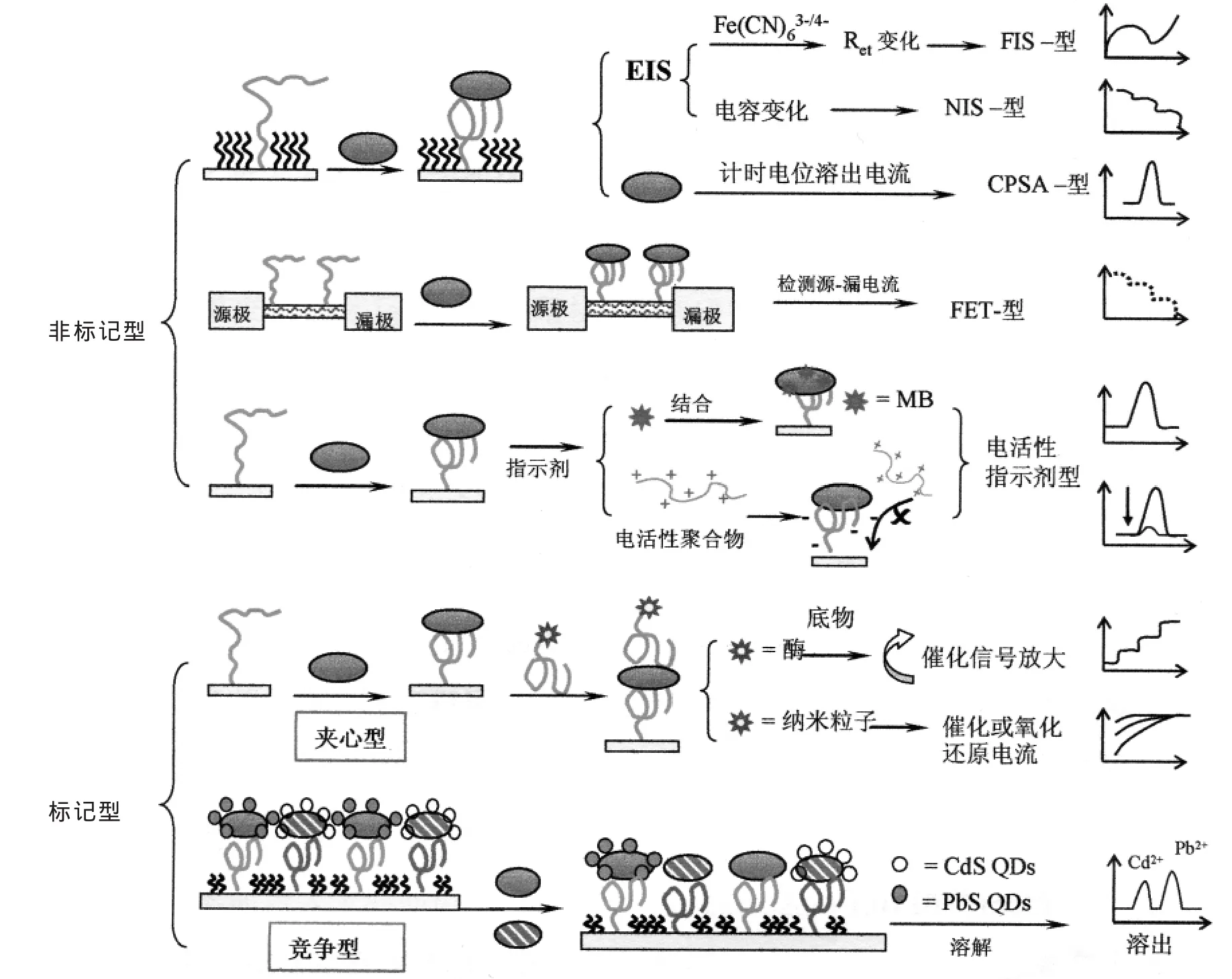
图1 亲和型核酸适体电化学传感器原理Fig.1 Electrochemical aptasensor based on affinity principle
有些实际操作情况下不能加入氧化还原探针。这样,电极/溶液界面就像是平行板电容器,电容(CSS)成为最重要的参数。因此,可利用非法拉第阻抗谱(NIS)研究核酸适体与配体的结合。Cui等报道了第一例利用NIS核酸适体传感器来检测血小板生长因子[14]。他们将PDGF-BB的核酸适体共价固定到氨基化的硅表面,随着PDGFBB的加入,几秒之后,电容线性降低,检出限为40 nmol/L。由于NIS技术不需要加入任何试剂,这种核酸适体传感器可用于体内监测。
1.1.2 计时电位溶出型(CPSA)核酸适体传感器
利用蛋白质固有的电活性也可以直接检测蛋白质。Wang的小组十年前就报道了利用CPSA跟踪肽和蛋白质的灵敏方法[15]。他们首次将核酸适体修饰的磁珠与CPSA相结合,构建了无标记化学法检测溶菌酶[16]。核酸适体与溶菌酶形成复合物,经分离后采用碱处理,使溶菌酶从磁珠上释放出来,分离出磁珠,剩下的溶菌酶利用酪氨酸和色氨酸残基的氧化进行CPSA分析,检测下限可达 350 fmol(7 nmol/L)。
1.1.3 场效应晶体管型(FET)核酸适体传感器
场效应晶体管转化器是构建电化学核酸适体传感器的另一种方法。在源极和漏极之间的通道固定传感元件是制备FET生物传感器的关键。为检测源-漏电流的变化,固定化传感层的厚度一般都要比德拜距离小[17~19],否则识别后带电荷的蛋白被相反电荷的离子所屏蔽,检测不到信号。抗原-抗体体系则不适用于这种类型适体传感器的构建。在盐溶液中抗体典型尺寸约为10~15 nm,那么抗原结合将超过德拜距离(3 nm,10 nmol/L离子浓度),无法检测到信号。而核酸适体的尺寸很小(约为1~2 nm),故适用于FET传感器的设计,识别过程很容易在双电层距离内进行并被检测到。Lee等首次报告了用于凝血酶检测的单壁碳纳米管场效应晶体管(SWNT-FET)核酸适体传感器[20]。SWNT-FET大多是采用标准化学气相沉积技术制备的,凝血酶核酸适体是被共价结合在改性的碳纳米管上。这种传感器响应迅速,检测限为10 nmol/L。Maehashi和Tamiya报道了另一种基于适体的碳纳米管场效应晶体管(CNT-FETs)传感器,对IgE的检测下限为250 pmol/L[21]。
1.1.4 吸附电活性指示剂型核酸适体传感器
利用电化学活性指示剂,如电活性小分子或离子聚合物,也可设计非标记型适体传感器。Hianik等利用亚甲基蓝(MB)作为电活性指示剂来检测核酸适体与凝血酶的相互作用。MB分子可以很容易地吸附到凝血酶上,MB的电信号响应就可以直接指示凝血酶的浓度,检测限为10 nmol/L[22]。
从静电作用方面来考虑,修饰核酸适体的电极带负电,如果加入带正电的聚合物探针,通过静电引力可以使其结合到电极表面。如果有目标蛋白质存在,蛋白质-核酸适体的结合会阻碍阳离子聚合物靠近电极表面,因此,骨架上带有电活性官能团的离子聚合物是可以检测电极表面核酸适体与目标蛋白相互作用的。 Leclerc等报道了利用2,4-二茂铁阳离子取代的聚噻吩作为电活性指示剂来直接检测凝血酶的方法[23]。该研究采用方波伏安法(SWV)来检测,电极表面有凝血酶结合时,二茂铁的电流信号被抑制,但没有做定量分析。
1.2 标记型电化学核酸适体传感器
核酸适体的靶物质如果是小分子,如腺苷等,适体结合时会将其包埋于特定的立体构象袋中,不能再结合其它适体。靶物质是蛋白质等大分子时,就可能含有两个以上结合适体的活性位点。对于后者,可以采用类似于免疫分析的策略,如夹心法和竞争法来设计标记型电化学核酸适体传感器。
1.2.1 酶标夹心型核酸适体传感器
夹心法广泛应用于免疫分析中。对于蛋白质分析,夹心法是一种理想的选择,因为目标蛋白分析物不需要标记。某些蛋白质,如凝血酶,存在多个结合位点可以和不同的特异性核酸适体结合。这就使得夹心型核酸适体传感器的设计成为可能。捕获核酸适体探针被固定在电极表面,检测适体探针上标记酶,用于电催化信号放大。Ikebukuro等首次报道了利用两种不同的凝血酶作为核酸适体的夹心型电化学适体传感器[24~25]。核酸适体Ⅰ固定在金电极上用于识别,葡萄糖脱氢酶(GDH)标记的核酸适体Ⅱ用于产生电信号。结果显示,采用对氧气不敏感的((PQQ)GDH)标记比GDH标记更灵敏,检测限为10 nmol/L,而用GDH作标记检测限为1 μmol/L。在夹心体系中,能够产生放大电信号的多功能标记物非常适用。Katakis等利用辣根过氧化酶标记的核酸适体发展了用于凝血酶检测的夹心型适体电化学传感器[26]。然而,由于酶标适体非特异性吸附比较强,检测限比较高(80 nmol/L)。
1.2.2 纳米粒子标记夹心型核酸适体传感器
金属纳米粒子(NP)是另一种能够催化电化学信号放大的标记物,它也被用于夹心型核酸适体电化学传感器的设计。Willner小组报道了铂纳米粒子标记的核酸适体用于夹心型传感器的设计,用于凝血酶的分析[27]。但他们的研究中,用于捕捉和检测的核酸适体是同一种。夹心反应后,铂纳米粒子催化H2O2还原产生催化电流。由于凝血酶层产生的高电子转移电阻,催化电流只能在极低的扫描速率下(20 mV/s)观察到,检测限在1 nmol/L左右。Fang的小组基于磁性纳米粒子标记的核酸适体和金纳米粒子标记的核酸适体,设计了一种灵敏的用于凝血酶检测的电化学生物传感器[28]。夹心反应后,采用磁场将免疫复合物(磁性纳米粒子标记适体/凝血酶/金标适体)富集纯化。在盐酸溶液中,Au纳米粒子在高电位下被氧化成Au(Ⅲ),再采用差分脉冲伏安法(DPV)检测还原电流。此方法灵敏度高,检出限可达1.42 pmol/L。
1.2.3 量子点(QD)标记竞争型核酸适体传感器
将电化学溶出测量与量子点示踪相结合已经被证实是一种灵敏有效方法用于蛋白质的生物分析。利用量子点电化学编码来做信号增强的复杂蛋白质分析近年来引起了广泛的研究兴趣。Wang等设计了竞争型多分析物电化学核酸适体传感器用于凝血酶和溶菌酶的同时检测[29]。首先,将巯基核酸适体及其相应的量子点标记蛋白固定在金表面上。然后加入待分析蛋白样品来取代标记的蛋白。洗涤后,将剩下的CdS和PbS量子点溶解,在修饰的玻碳电极上进行溶出伏安分析。由于量子点的有效放大作用,可以获得亚皮摩尔级的检测限。与传统的抗原-抗体免疫检测相比,量子点电化学核酸适体传感器更加灵敏,这可能得益于标记蛋白与紧密修饰核酸适体的电极表面间低的亲合力。
2 构型变换型适体生物传感器
当核酸适体与特定的蛋白质结合时,有典型的构型变换,而抗体则没有这样的特性。这个性质可以被用来设计构型变换型适体传感器,原理如图2所示。
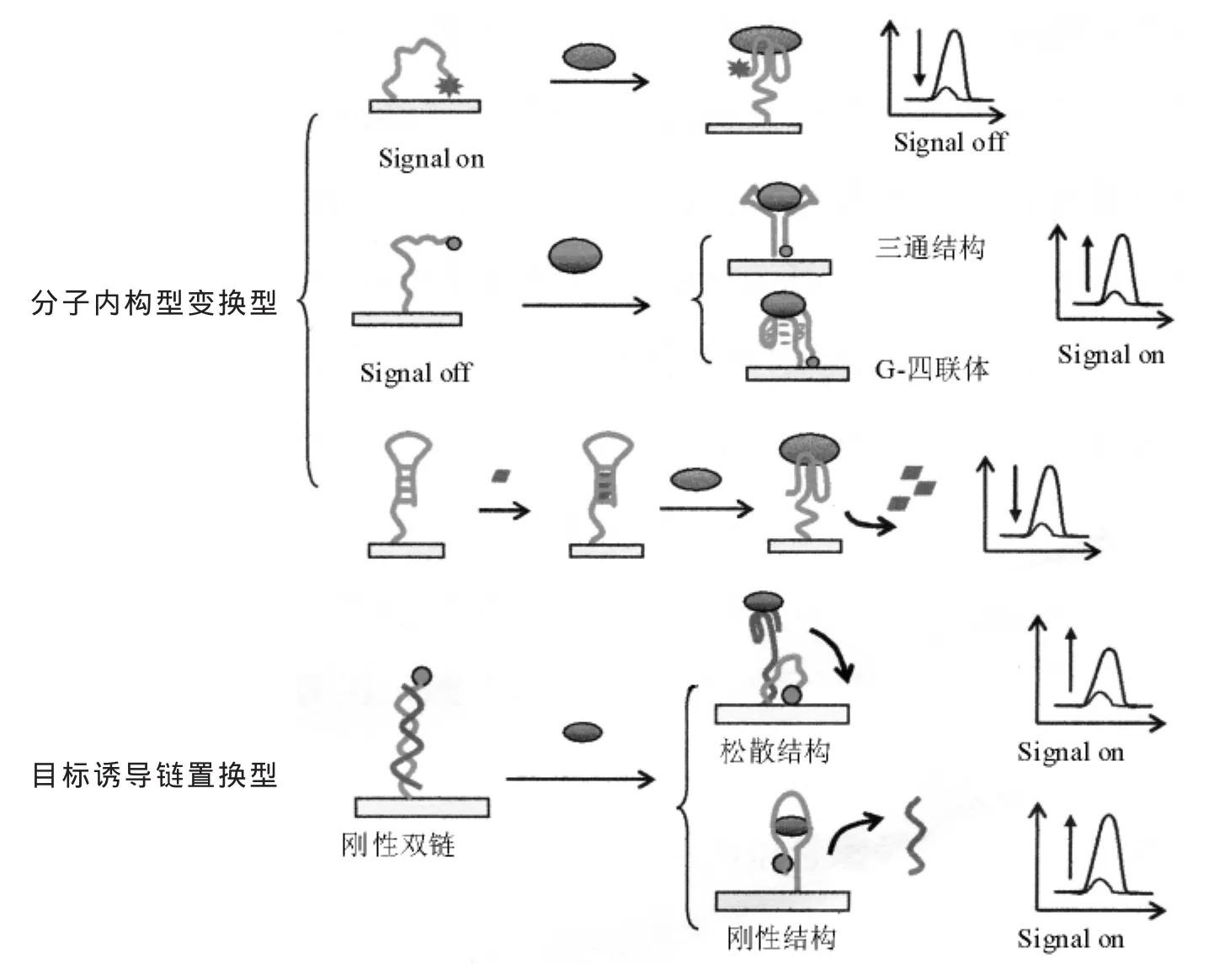
图2 构型变换型核酸适体电化学传感器原理Fig.2 Electrochemical aptasensor based on Conformation change principle
2.1 分子内构型变换型
1996年首次报道了荧光分子信标作为无标记无试剂型探针用于目标DNA分析[30~32]。分子信标包含一个用于信号产生的茎部和一个用于与核酸互补杂交的环部。加入目标寡核苷酸,随着杂化的进行,结合的茎结构打开并自发的产生构型变换。这种构型变换是合理的,因为杂化时产生的吉布斯自由能降低比茎部结合时要大。这种构型变换已被用于设计各种淬灭和荧光共振能量转移(FRET)型分析[33~34]。 已有不少研究者在探索将环部采用适体序列取代,发展类似荧光原理的基于适体信标的电化学传感器。通常,将适体信标的一端修饰官能团用于在电极表面的固定化,另一端修饰电活性标记物来产生电信号。标记物的氧化还原反应是距离决定的。识别后,分子内构型变换改变了电化学标记物和电极表面的距离,产生可测的信号改变[35~36]。这种构型变换既可以设计信号减弱(signal-off)型,也可以设计信号增强型(signal-on)适体传感器。
在之前的研究中,Plaxco小组使用标记有MB的巯基化32-碱基凝血酶适体设计了一种无标记signal-off型电化学适体传感器检测血清中的凝血酶(2005年7月)[37]。在结合前,适体保持非折叠构型,使MB分子碰撞电极表面并转移电子。加入凝血酶,信标适体折叠成G-四联体结构以利于凝血酶-适体间发生适应性结合作用。由于靶物质的结合引起标记物与电极距离增加,所以电子转移受到抑制。这种电化学适体传感器的关键优势是宽的线性响应范围(从纳摩尔到微摩尔)和能够实现血清样品中凝血酶的灵敏检测。这种基于靶物质的结合引起构型变换的电化学适体传感器,提供了一种无试剂、再生和灵敏的方法用于复杂样本中目标分析物的检测。但signal-off型适体传感器也有其局限性。目标分析物的结合减小了而不是增大了电化学信号。信号增溢是受限制的,因为最多只有100%的信号被抑制。并且,污染物以及其它物质所产生的“假信号”与特异靶物质的结合所产生的信号很难区别。上述难题采用signal-on型适体传感器很容易解决,因为靶物质的识别会带来较大的信号增强。
Radi等报道了一种用二茂铁作为氧化还原标记物的巯基化15-碱基凝血酶适体用于研究,获得了与Plaxco不同的signal-on结果[38]。主要原因是适体碱基长度的不同和阻断分子巯基乙醇的引入。他们认为信号增强是凝血酶的结合引起了从随机卷曲构型到四联体结构的分子内构型变换。形成四联体后,二茂铁向电极表面靠近,氧化电流增强。
还有些适体与靶物质结合后形成三维空间结构,而靶物质不存在的情况下只与其中一个茎相接触,仍部分地保持未折叠。Plaxco小组利用构型变换设计了检测可卡因和PDGF-BB的电化学适体传感器。他们利用MB标记的巯基化适体部分的未折叠结构在电极表面形成自组装单分子层[39]。当加入目标分析物可卡因,适体很快折叠成与可卡因结合的三维结构,减小了MB标记物与电极表面的距离,峰电流增大,此方法的检测限为10 μmol/L。在80 s内就可以获得97%的变化,4 min内全部完成。该传感器在室温下简单的冲洗就能再生。另一种PDGF-BB适体传感器[40],在目标物存在时适体也变换成稳定的三维结构,使MB标记物接近电极表面。这种传感器可在未稀释的血清中1 nmol/L PDGF-BB和50%血清中50 pmol/L PDGF-BB的直接检测。这种基于分子内构型变换的电化学适体传感器提供了一些有效的方法,来解决采用荧光分子信标测试临床样品时存在的高背景干扰问题。基于分子内构型变换的信标适体电化学传感器背景干扰要比荧光小很多,有望在临床分析中得到应用。
适体上富含鸟嘌呤而形成紧密的G-四联体结构。 这种结构在一价阳离子特别是钾离子中能稳定存在。也就是说,钾离子与富G适体有特异结合能力,并能够促进适体从松散的卷曲结构到稳定的G-四联体结构转变。这就使设计钾离子型电化学适体传感器成为可能。Radi等首先报道了基于构型变换的钾离子选择性识别的电化学适体传感器[41]。检测限约为0.015 mmol/L。
一些平面染料分子可以插入双链DNA中并经DNA碱基对π堆积实现电子转移而被氧化。这个原理也被用于设计信标适体传感器用于靶分子的检测。Kim等使用MB作为电化学标记物提出了基于信标适体的电化学检测方法[42]。共价固定的信标适体呈发卡结构,这样MB分子就可以插入到茎部双链中,当遇到目标物凝血酶,信标产生构型变换,凝血酶与环部结合打开结合的茎部,并释放MB分子,引起MB峰电流的减小,检测限为11 nmol/L。
2.2 目标诱导链置换型
Plaxco等将“目标诱导链置换”策略引入到电化学适体传感器的设计中[43]。在金电极上组装巯基化DNA适体的单分子层,该DNA适体链包含一段15个碱基的凝血酶适体和一段适体与巯基间的连接序列。然后,加入既能与适体片段又能与连接序列杂化的MB标记的寡核苷酸。形成的刚性双链结构能够阻止MB与电极表面接触。加入凝血酶后,适体片段折叠成G-四联体结构结合凝血酶,释放出标记MB的寡核苷酸片段,产生易松散的链,使MB标记物接近电极表面,产生电流,其检测限是3 nmol/L。但该传感器不可再生。Wu等在一个类似的研究中报道了腺苷诱导链置换,使二茂铁标记的适体从传感界面上分离出来,设计了一种signal-off型电化学适体传感器[44]。
多数基于构型变换的适体传感器的设计总有一种状态是松散结构要么是 “on”要么是“off”状态。在这种松散结构中,电化学信号是通过末梢的标记物与电极表面动态碰触产生的,这就使信号响应受到限制。如果能够设计构型明确的“on”或“off”型刚型结构,就能克服这些缺点。Fan等设计了检测三磷酸腺苷(ATP)的电化学适体传感器[45],他们使用的长链DNA由适体片段、双链部分和阻断部分组成,适体片段用于高亲和性ATP识别,与互补链杂化的双链部分用于支撑结构。标记3'-SH和5'-二茂铁的DNA适体首先以杂交双链形式自组装到金表面。在这种刚性的长结构中,二茂铁的电子转移效率低。ATP的加入使双链结构破坏,并使互补DNA链解开。这样,由于刚性的ATP-适体三级结构的形成促使二茂铁标记物靠近电极表面,产生可测电流。由于杂交双链结构和三级结构都是刚性的,适体传感器具有明确的“signal-off”和“signal-on”结构状态,检测限可以覆盖nmol/L到mmol/L范围。
3 酶参与型
适体与目标蛋白质的特异结合将改变一些酶反应的特性,例如核酸酶的水解反应和等温滚环扩增(RCA)反应等。这些反应特性可以被用来设计一些基于酶参与型的适体传感器,原理如图3所示。
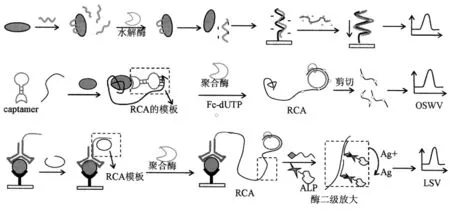
图3 酶参与型核酸适体电化学传感器原理Fig.3 Electrochemical aptasensor based on enzyme-participated strategies
适体是合成寡核酸或多肽分子,很容易被核酸酶水解。但有目标蛋白质存在时,形成的适体-蛋白质复合体会阻止适体的降解。Leclerc等利用这种特性设计了一种基于适体检测的凝血酶电化学适体传感器。该研究使用了与适体互补的单链肽核酸(PNA)和阳离子噻吩高聚物[23]。特异的适体首先加入到凝血酶溶液中形成适体-凝血酶复合体。然后,加入核酸酶水解溶液中剩余的适体,只留下键合的适体。在95℃下处理10 min,使蛋白质变性,结合的适体被释放到溶液中。滴加1 μL该溶液到中性PNA探针修饰的电极上实现检测。PNA-适体杂化后,电极表面带负电荷。将其浸泡在阳离子高聚物中,通过静电作用将阳离子聚合物吸附到电极上,形成PNA-适体-高聚物复合体,产生“signal on”型电化学响应,实现了凝血酶的定量分析,检测下限是75 nmol/L。
环状 DNA 适体(“captamers”)是将两种或更多折叠适体以及核酸双链或联结结构包含在一个环状分子中的DNA[46~47]。适体处于联结结构的顶端,可以提供多个蛋白质结合活性位点。与传统的适体相比,这种环状DNA适体具有更高的热稳定性和更好的耐核酸酶降解特性。Captamer正如环状DNA一样可以用作RCA反应的模板。King等采用环状DNA适体设计了聚合酶介导的临近延伸 “proximity extension”法用于蛋白质的定量检测[48]。该方法采用了一个哑铃状Captamer和一条线性适体,它们各含有一个凝血酶的识别位点,当有目标物存在时,Captamer和线性适体的一端像钳子一样将凝血酶夹住,线性模板的另一端与Captamer的环状顶端可以杂交,提供后继酶介延伸和等温扩增的引物。Captamer就成为随后等温扩增的模板。当有凝血酶存在时,通过酶介扩增反应,产生环状模板序列循环复制的长链高分子产物。将产物设计成含有限制性内切酶的识别位点,就可以将其切割成期望长度的DNA片断。为了实现电化学检测,大约35%的扩增反应的核苷酸采用电活性核苷酸取代 (vinyl-FcdUTP)。该电活性DNA片断能够被互补链捕获到电极表面,采用方波伏安法(OSWV)检测,线性响应范围从pmol/L到nmol/L。
楚等提出了另一种基于免疫-RCA的适体电化学传感器用于PDGF-BB的超灵敏检测[49]。在电极表面形成一种像抗体-PDGF-BB-适体-引物复合体这样的三明治结构后,引入一种与引物结合的“C”形挂锁探针,该探针提供了后续RCA反应的模板,在DNA聚合酶和dNTPs存在时,发生等温扩增产生长链RCA产物,该产物的电化学检测与King等不同之处在于引入了生物素标记的检测探针和亲和素标记的ALP酶,利用生物素-亲和素的特异结合作用和探针杂交,将ALP酶结合到RCA产物上,ALP酶催化银沉积,再采用溶出伏安法检测。该方法结合了滚环扩增和酶催化金属沉积两步放大,实现了超灵敏的选择性检测,检测范围跨越4个数量级,下限可达10 fmol/L。黄等采用类似原理将PDGF-BB采用抗体和适体-引物捕获到电极表面[50],然后引入的是一个单链环形质粒DNA,在聚合酶作用下延伸形成环形双链DNA,通过嵌入亚甲基蓝分子产生电化学响应,该传感器检测下限为18 pg/mL。
4 表面临近杂交型
Fredriksson等2002年在 Nature Biotechnology上发表了基于邻近分析检测蛋白质的研究[51],首次提出了邻近分析的概念。邻近分析是指一对亲和探针同时识别目标分子并与目标分子高亲和力的结合,形成了一对邻近的探针,从而使检测信号增强。这些报道大多是溶液的均相反应,蒋等将这一概念应用到适体传感器的设计,提出了表面临近杂交型电化学适体传感器[52],原理如图4所示。一对相同的核酸适体同时识别以二聚体形式存在的PDGF-BB后,由于临近效应使适体探针两尾端序列同时与电极表面固定的两条短链DNA杂交,适体末端修饰了二茂铁,这种临近杂交促使二茂铁充分靠近电极表面,产生显著的氧化还原电流。该传感器响应范围可从1.0 pg/mL到20 ng/mL,检测限可达1.0 pg/mL。他们还将表面邻近杂交分析技术推广应用到核酸分子靶标的电化学传感器构建中[53]。

图4 表面临近杂交型核酸适体电化学传感器原理Fig.4 Electrochemical aptasensor based on proximity-dependent surface hybridization principle
总的来说,虽然研究者们已经探索了多种构建核酸适体电化学传感器的方法,并获得了较满意的分析性能,但适体电化学传感器的研究仍处于它的初级阶段,还有很多重要问题值得深入研究。理论上任何目标物的适体都能够合成和应用,但实际上只有少数蛋白质和小分子的核酸适体得到研究。随着SELEX技术的发展,更多具有高亲和力和低成本的适体可以合成出来,作为抗体的替代物或补充用于适体生物传感器的构建。目前,对电化学适体传感器的分析性能和各项参数,如选择性、特异性、重现性、稳定性和线性响应的研究和评价都不充分,在实际体系中的应用也还需要拓展。因此,今后电化学核酸适体电化学传感器的发展还是在以下几个方面:利用纳米生物新技术发展新的信号扩增方法,获得超灵敏的信号响应;探索新的检测原理和分析技术,发展操作简便可控,稳定性和重现性好的通用型新方法;寻找对更多靶分子具有高亲和力的核酸适体,在免疫分析难以解决的研究领域拓展核酸适体传感器的应用,解决一些具体问题;建立基于电化学核酸适体的高通量分析技术;利用电化学传感器的优势,发展能自动化和实时监测的分析新技术。
[1]Robertson D L,Joyce G E.Selection in vitro of an RNA enzyme that specifically cleaves single-stranded DNA[J].Nature,1990,344(6 265):467~468.
[2]Ellington A D,Szostak J W.In vitro selection of RNA molecules that bind specific ligands[J].Nature,1990,346(6 287):818~822.
[3]Tuerk C,Gold L.Systematic evolution of ligands by exponential enrichment:RNA ligands to bacteriophageT4DNA polymerase[J].Science,1990,249(4 698):505~510.
[4]Nimjee S M,Rusconi C P,Sullenger B A.Aptamers:An Emerging Class of Therapeutics[J].Annual Review of Medicine,2005,56:555~583.
[5]Jayasena S D.Aptamers:an emerging class of molecules that rival antibodies in diagnostics[J].Clinical Chemistry,1999,45(9):1 628~1 650.
[6]Camille L A Hamula,Jeffrey W Guthrie,Hongquan Zhang,et a1.Selection and analytical applications of aptamers[J].Trends in Analytical Chemistry(TrAC),2006,25(7):681~691.
[7]Herman T,Patel D J.Adaptive Recognition by Nucleic Acid Aptamers[J].Science,2000,287(5 454):820~825.
[8]Noeske J,Buck J,Furtig B,et a1.Interplay of‘induced fit’and preorganization in the ligand induced folding of the aptamer domain of the guanine binding riboswitch[J].Nucleic Acids Research,2007,35(2):572~583.
[9]Rodriguez M C,Kawee A,Wang J.Aptamer biosensor for label-free impedance spectroscopy detection of proteins based on recognition-induced switching of the surface charge[J].Chem.Commun.,2005,34:4 267~4 269.
[10]Xu D K,Xu D W,Yu X B,et a1.Label-Free Electrochemical Detection for Aptamer-Based Array Electrodes[J].Anal.Chem.,2005,77(16):5 107~5 113.
[11]Radi A E,Acero Sanchez J L,Baldrich E,et a1.Reusable Impedimetric Aptasensor[J].Anal.Chem.,2005,77(19):6 320~6 323.
[12]Cai H,Lee T M H,Hsing I M.label-free protein recognition using an aptamer-based impedance measurement assay[J].Sensors and Actuators B:Chemical,2006,114(1):433~437.
[13]Xu Y,Yang L,Fang,Y Z,et a1.An aptamer-based protein biosensor by detecting the amplified impedance signal[J].Electroanalysis,2006,18(15):1 449~1 456.
[14]Liao W,Cui X T.Reagentless aptamer based impedance biosensor for monitoring a neuro-inflammatory cytokine PDGF[J].Biosensors and Bioelectronics,2007,23(2):218~224.
[15]Cai X,Wang J,Palecek E,et a1.Potentiometric stripping analysis of bioactive peptides at carbon electrodes down to subnanomolar concentrations[J].Anal.Chim.Acta.,1996,332(1):49~57.
[16]Kawde A,Rodriguez M,Wang J,et a1.Label-free bioelectronic detection of aptamer-protein interactions[J].Electrochem.Commun,2005,7(5):537~540.
[17]Park K Y,Kim M S,Choi S Y.Fabrication and characteristics of MOSFET protein chip for detection of ribosomal protein[J].Biosensors and Bioelectronics,2005,20(10):2 111~2 115.
[18]Matsuo T,Wise K D.An integrated field effect electrode for biopotential recording [J].IEEE Transactions on Biomedical Engineering,1974,BME-21(6):485~487.
[19]Michael J S,Arshak P.Recent advances in biologically sensitive field-effect transistors[J].Analyst,2002,127:1 137~1 151.
[20]So H M,Kim H,Lee J O,et a1.Single-walled carbon nanotube biosensorsusing aptamersas molecular recognition elements[J].J.Am.Chem.Soc,2005,127(34):11 906~11 907.
[21]Maehashi K,Matsumoto K,Tamiya E,et a1.Label-Free Protein Biosensor Based on Aptamer-Modified Carbon Nanotube Field-Effect Transistors[J].Anal.Chem,2007,79(2):782~787.
[22]Hianik T,Ostatná V,Zajacová Z,et a1.Detection of aptamer-protein interactions using QCM and electrochemical indicatormethods[J].Bioorganic&Medicinal Chemistry Letters,2005,15(2):291~295.
[23]Fabien Le Floch,Hoang A Ho,Mario Leclerc.Label-Free Electrochemical Detection of Protein Based on a Ferrocene-Bearing Cationic Polythiophene and Aptamer[J].Anal.Chem.,2006,78(13):4 727~4 731.
[24]Ikebukuro K,Kiyohara C,Sode K.Novel electrochemical sensor system for protein using the aptamers in sandwich manner[J].Biosens Bioelectron,2005,20(10):2 168~2 172.
[25]Ikebukuro K,Kiyohara C,Sode K.Electrochemical detection of protein.using a double aptamer sandwich[J].Anal.Lett.,2004,37(14):2 901~2 909.
[26]Mir M,Vreeke M,Katakis I.Different Strategies To Develop an Electrochemical Thrombin Aptasensor[J].Electrochemical Communication,2006,8(3):505~511.
[27]Polsky R,Gill R,Willner I,et a1.Nucleic Acid-Functionalized Pt Nanoparticles:Catalytic Labels for the Ampli-fied Electrochemical Detection of Biomolecules[J].I.Anal.Chem.,2006,78(7):2 268~2 271.
[28]Zheng J,He P G,Fang Y Z,et a1.Study on an electrochemical biosensor for thrombin.recognition based on aptamers and nano particles[J].Science in China,B,2006,36(6):485~492.
[29]Hansen J A,Wang J,Kawde A N,et a1.Quantum-Dot/Aptamer-Based Ultrasensitive Multi-Analyte Electrochemical biosensor[J].J.Am.Chem.Soc.,2006,128(7):2 228~2 229.
[30]Tyagi S,Kramer F R.Molecular beacons:probes that fluoresce upon hybridization[J].Nat Biotechnol,1996,14(2):303~308.
[31]Tyagi S,Bratu D P,Kramer F R.Multicolor molecular beacons for allele discrimination[J].Nature Biotechnology,1998,16:49~53.
[32]Fang X,Li J J,Perlette J,et a1.Molecular beacons:novel fluorescent probes[J].Anal Chem,2000,72:747A ~753A.
[33]Tan W H,Wang K M,Drake T J.Molecular beacons[J].Current Opinion in Chemical Biology,2004,8(5):547~553.
[34]Santangelo P J,Nix B,Tsourkas A,et a1.Dual FRET molecular beacons for mRNA detection in living cells[J].Nucleic Acids Research,2004,32(6):e57/1~e57/9.
[35]Nutiu R,Li Y.Structure-switching signaling aptamers[J].J Am Chem Soc,2003,125(16):4 771~4 778.
[36]Nutiu R,Li Y.In vitro selection of structure-switching signaling aptamers[J].Angew Chem Int Ed Eng,2005,144:1 061~1 065.
[37]Arica A,Heeger A J,Plaxco K W,et a1.Label-Free Electronic Detection of Thrombin in Blood Serum by Using an Aptamer-Based Sensor[J].Angew.Chem.Int.Ed.,2005,44(34):5 456~5 459.
[38]Radi A E,Baldrich E,O'Sullivan C K,et a1.Reagentless,reusable,ultrasensitive electrochemical molecular beacon aptasensor[J].J.Am.Chem.Soc,2006;128(1):117~124.
[39]Baker B R,Lai R Y,Plaxco K W,et a1.An electronic,aptamer-based small-molecule sensor for the rapid,label-free detection of cocaine in adulterated samples and biological fluids[J].J.Am.Chem.Soc,2006,128(10):3 138~3 139.
[40]Lai R Y,Plaxco K W,Heeger A J.Aptamer-Based Electrochemical Detection of Picomolar Platelet-Derived Growth Factor Directly in Blood Serum[J].Anal.Chem.,2007,79(1):229 ~233.
[41]Radi A E,O'Sullivan C K.Aptamer conformational switch as sensitive electrochemical biosensor for potassium ion recognition[J].Chem Commun,2006,28(32):3 432~3 434.
[42]Bang S G,Cho S H,Kim B G.A novel electrochemical detection method for aptamer biosensors[J].Biosensors and Bioelectronics,2005,21(6):863~870.
[43]Xiao Y,Piorek B D,Plaxco K W,et a1.A reagentless signal-on architecture for electronic,aptamer-based sensors via target-induced strand displacement[J].J Am Chem Soc,2005,127(51):17 990~17 991.
[44]Wu Z S,Guo M M,Zhang S B,et a1.Reusable Electrochemical Sensing Platform for Highly Sensitive Detection of Small Molecules Based on Structure-Switching Signaling Aptamers[J].Anal.Chem.,2007,79(7):2 933~2 939.
[45]Zuo X L,Song P P,Fan C H,et a1.A Target-Responsive Electrochemical Aptamer Switch(TREAS)for Reagentless Detection of Nanomolar ATP[J].J.Am.Chem.Soc.,2007,129(5):1 042~1 043.
[46]Di Giusto D A,King G C.Construction,stability,and activity of multivalent circular anticoagulant aptamers[J].J Biol Chem,2004,279(45):46 483~46 489.
[47]King G C,Knox S M,Williams B B,et a1.Multivalent Circular Aptamers:Versatile Nanostructures for Biomedical Applications[J].NIST-Nanotech,2005,1:258~261.
[48]Di Giusto D A,Wlassoff W A,Gooding J J,et al.Proximity extension of circular DNA aptamers with real-time protein detection[J].Nucleic Acids Res,2005,33(6):e64.
[49]Zhou L,Ou L J,Chu X,et a1.Aptamer-Based Rolling Circle Amplification:A Platform for Electrochemical Detection of Protein[J].Anal.Chem,2007,79(19):7 492 ~7 500.
[50]Huang Y,Nie X M,Gan S L,et a1.Electrochemical Immunosensor Based on Aptamer Primed Polymerase Amplification[J].Analytical Biochemistry,2008,382(1):16~22.
[51]Fredriksson S,Gullberg M,Jarvius J,et al.Protein detection using proximity-dependent DNA ligation assays[J].Nature Biotechnology,2002,20(5):473~477.
[52]Zhang Y L,Huang Y,Jiang J H,et al.Electrochemical Aptasensor Based on Proximity-Dependent Surface Hybridization Assay for Single-Step,Reusable,Sensitive Protein Detection[J].Journal of the American Chemistry Society,2007,129(50):15 448~15 449.
[53]Zhang Y L,Huang Y,Jiang J H,et al.Electrochemical DNA Biosensor Based on the Proximity-Dependent Surface Hybridization Assay[J].Analytical Chemistry,2009,81(5):1 982~1 987.
猜你喜欢
中国慈善家(2022年3期)2022-06-14
现代苏州(2022年9期)2022-05-26
快乐语文(2021年34期)2022-01-18
成都医学院学报(2021年2期)2021-07-19
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08
中国(俄文)(2020年8期)2020-11-23
天然产物研究与开发(2018年10期)2018-11-06
北京航空航天大学学报(2017年10期)2017-04-20
航天返回与遥感(2014年4期)2014-07-31
食品工业科技(2014年5期)2014-03-11
