
小鼠胚胎干细胞药物代谢酶CYP3a11基因敲除的实验研究
2010-06-08 10:31曾军王雪丁陈宏远黄民杜军
中国医药生物技术 2010年4期
曾军,王雪丁,陈宏远,黄民,杜军
细胞色素酶 P450 3a(CYP3a)负责许多内源性和异生化合物的氧化代谢,是人体肝脏和小肠最丰富的 CYPs,参与毒素、致癌物质、胆汁酸、类固醇激素以及超过 50%临床用药等的初级代谢。CYP3a11 是小鼠肝内微粒体中优势表达的 CYP3a基因[1],在功能上与人类的 3a4 相类似,但 CYP3a的药物代谢谱存在着明显的种系差异[2],这给动物药物实验的有效性带来挑战。利用基因敲除技术构建药物代谢酶基因缺陷型小鼠和人源化小鼠,可为深入研究药物代谢酶的功能、药效学和药物致癌等提供有效模型[3]。基于此,我们设计并构建了小鼠药物代谢酶 CYP3a11 基因敲除载体,并对小鼠E14 胚胎干细胞(embryo stem cell,ES cell)的CYP3a11 基因进行了敲除,为进一步制作 CYP3a11基因缺陷型小鼠作准备。
1 材料与方法
1.1 材料
1.1.1 质粒、菌株和 ES 细胞株 质粒 pBluescript II SK+phagemid 为美国 Stratagene 公司产品;pCR2.1-TOPO 为美国 Invitrogen 公司产品;大肠杆菌 DH5α 由本室保存;129 品系小鼠 E14 ES细胞株为中科院上海生化与细胞研究所提供;ES细胞培养及筛选所需要的支持细胞来自其他基因敲除小鼠孕 12.5 d 原代培养的胚胎成纤维细胞(mouse embryo fibroblast,MEF)。
1.1.2 工具酶、试剂盒及其他试剂 所有引物均为上海生工生物工程技术服务有限公司产品;限制性内切酶、T4DNA 聚合酶、T4DNA 连接酶、Taq酶和 PfuUltra™ 酶分别购自美国 Promega、NEB、Stratagene 公司;pfu T/A 克隆试剂盒为上海生物工程公司产品;QIAEX II 凝胶回收试剂盒为德国QIAGEN 公司产品;Ganc 和 G418 sulfate 购自美国 Gibco 公司;α-32P-dCTP 为美国 Amersham 公司产品;标记探针用缺口平移试剂盒是华美生物工程公司产品;1 kb ladder DNA plus 核酸分子量标记购于美国 Invitrogen 公司;Defined 胎牛血清购自美国 Hyclone 公司;重组小鼠白血病抑制因子(Leukemia inhibitory factor,LIF)购自美国Millipore 公司;牛蛋白胨、琼脂糖试剂分别购自美国 Sigma 和 Biorad 公司;其他化学试剂均为分析纯。
1.2 方法
1.2.1 载体构建方法 取培养的 E14 ES 细胞株,裂解并提取基因组 DNA。根据 NCBI 公布CYP3a11 序列[GeneID:13112,Locus tag:88609]设计引物如下:
P1:5’ AGCTGGCCTGGATATAACTGTGTATA CCAG 3’,P2:5’ GGGCTAGCTTACCTTTATGAGA GACTTTGT 3’,此对引物的产物为敲除基因的长臂上游(site:1940-5326)3.3 kb 片段,命名为LA-up;
P3:5’ AAGCTAGCCCTTCCTGGTCTATAGA CAAGG 3’,P4:5’ CACTCGAGGCCTGGAAGAT GCTCAATTACT 3’,此对引物的产物为敲除基因的长臂下游(site:5317-9318)4 kb 片段,命名为LA-low;
P5:5’ GGAACCCATATGTGAGAGCGCAGAG AGACT 3’,P6:5’ AACCCGGGACTCAGGGCTAC ACACTGTAAT 3’,此对引物的产物为敲除基因的短臂(site:89-2051)2 kb 片段,命名为SA。
建立如下体系:模板基因组 DNA 0.1 μg,10×PCR buffer 2 μl,dNTP 20 μmol/L,引物 P1 和 P2各 500 nmol/L,PfuUltra™ 酶 3 U,补加 dd H2O至 20 μl。PCR 反应条件为:94℃变性 5min,再进行如下循环程序 30 次:94℃变性 30 s,56℃退火 30 s,72℃延伸 1min,最后 72℃延伸 8min。将 PCR 产物进行 0.8%琼脂糖凝胶电泳鉴定。
小鼠 CYP3a11 基因敲除载体的构建策略如图1 所示。扩增得到的 LA-up 和 LA-low 片段,依次连接到 pCR2.1-TOPO 载体中,将来源于质粒pNEOZTK 中的负筛选标记 tk 基因,插入至LA-low 的下游,扩增后做相应酶切并连接得到包含完整下游同源臂的 LA-tk-pCR2.1-TOPO 载体。将新霉素磷酸转移酶基因 PGK neo 片段连接到pBluescript II SK+载体的多克隆位点的EcoRI 及BamHI 之间,得到载体 neo-pBluescript II SK+。扩增得到的 SA 片段行ApaI +HindIII 酶切,所得0.65 kb 片段连接至ApaI +HindIII 消化的 neopBluescript II SK+载体中,位置在 PGK neo 基因的上游,得到包含上游同源臂的 SA-neo-pBluescript II SK+载体。NotI +ApaI 消化 SA-neo-pBluescript II SK+载体,获得ApaI-SA-neo-NotI片段,将其连接至 LA-tk-pCR2.1-TOPO 载体相应的酶切消化的位点,从而获得具有完整 SA-neo-LA-tk 的 CYP3a11基因敲除载体 pCR-CYP3a11_KO。

图1 小鼠 CYP3a11 基因敲除载体 pCR-CYP3a11_KO构建策略图Figure 1 Strategy for the construction of pCR-CYP3a11_KO,the CYP3a11 knock-out vector of mouse
1.2.2 小鼠 E14 ES 细胞的准备和转染 待培养皿中 MEF 长满,用 10 μg/ml 丝裂霉素处理 4 h后,接种 E14 ES 细胞在 MEF 上进行培养。至 ES克隆长满,收集细胞,计数,以 1×107个细胞/ml浓度重悬于电转缓冲液(20mmol/L 羟乙基哌嗪乙磺酸,pH 7.3,137mmol/L NaCl,5mmol/L KCl,0.7mmol/L Na2HPO4,0.6mmol/L 葡萄糖)0.1mmol/L 2-mercaptoethanol(2-ME),加至电极宽度为4mm 的电击杯。按每杯 20~30 μg 的量加入提纯的线性化的 pCR-CYP3a11_KO 载体(PvuI消化),将电击杯冰上预冷,并转移至电穿孔仪电转,电转参数为250 V/500 μF,电转后室温静置30min。然后将电转后的细胞均匀加入含丝裂霉素处理的 MEF 的培养皿中,至 37℃孵箱中培养。
1.2.3 CYP3a11 基因敲除 E14 ES 克隆的初步筛选和单克隆 24~36 h 后,除去电转染的 E14 ES 细胞培养液,加入含有 500 μg/ml G418 和0.2 μmol/L Ganc 的全培养液进行筛选,每 2~3 天换液 1 次,视情况加减 G418 筛选压力,4 周后,可见 ES 克隆增殖。6 周后挑取克隆转移到 24 孔板中进行单克隆培养,维持 100 μg/ml G418 的筛选压力,并逐渐扩大培养面积,用于冻存及提取DNA 进行鉴定。
1.2.4 CYP3a11 基因敲除 E14 ES 克隆的 PCR检测 将培养在 24 孔板中的 E14 ES 细胞进行传代时,取部分细胞(约 2×103~3×103个)用 PBS 洗涤并离心收集,置于 200 μl 裂解液(10mmol/L Tris-HCl,pH 8.0,5mmol/L EDTA,0.2%SDS,0.2mmol/L NaCl,0.1 mg/ml 蛋白酶中,37℃消化 3 h,离心,取 5 μl 上清液进行 PCR扩增。PCR 引物设计如下:
P7:5’ GGATTGTGAAACCATACCCA 3’,P8:5’ AAAGCGCATGCTCCAGACTG 3’,其中 P7 位于 SA 上,P8 位于 PGK neo 基因上游,预期产物为830 bp,PCR 扩增条件:94℃1min,58℃1min,72℃2min,35 个循环。扩增产物用 0.8%的琼脂糖凝胶进行电泳分析。
1.2.5 CYP3a11 基因敲除 E14 ES 克隆的Southern blot 检测 培养细胞基因组 DNA 的提取:将 PCR 检测为阳性的 E14 ES 克隆进行增殖培养和冷冻保存,当细胞数目达到约 1×107时提取 DNA。基因组 DNA 经电泳分析和光密度测定后,取 30 μg 进行HindIII 酶切,电泳分离后进行 Southern 转移。
探针的分离和标记:探针为1.6 kb 的 PGK neo。用缺口平移试剂盒进行同位素探针标记。反应体系为50 μl,包括 1 μg 的待标记 PGK neo。Southern 杂交按常规方法进行。
2 结果
2.1 CYP 3a11 基因敲除载体的酶切检测及线性化
基因敲除载体 pCR-CYP3a11_KO 经过ApaI、BamHI、BglII、EcoRV、NotI 酶切,检测结果如图2 所示,所有的片段都与预期大小一致。

图2 pCR-CYP3a11_KO 的酶切图谱Figure 2 Restriction enzymic analysis of the pCR-CYP3a11_KO
2.2 CYP 3a11 基因敲除的 E14 ES 细胞的初步筛选和单克隆的分离培养
转染后经 G418 筛选 2 周后,95%以上的E14 ES 细胞死亡。约 4 周左右可见阳性细胞克隆形成,如图3 所示。6 周左右挑取克隆,最后分离至 24 孔培养板培养的 E14 ES 单克隆共 97 个。

图3 G418 筛选 6 周后 E14 ES 细胞形成单克隆Figure 3 Single E14 ES cell colony formed under 6 weeks selection of G418
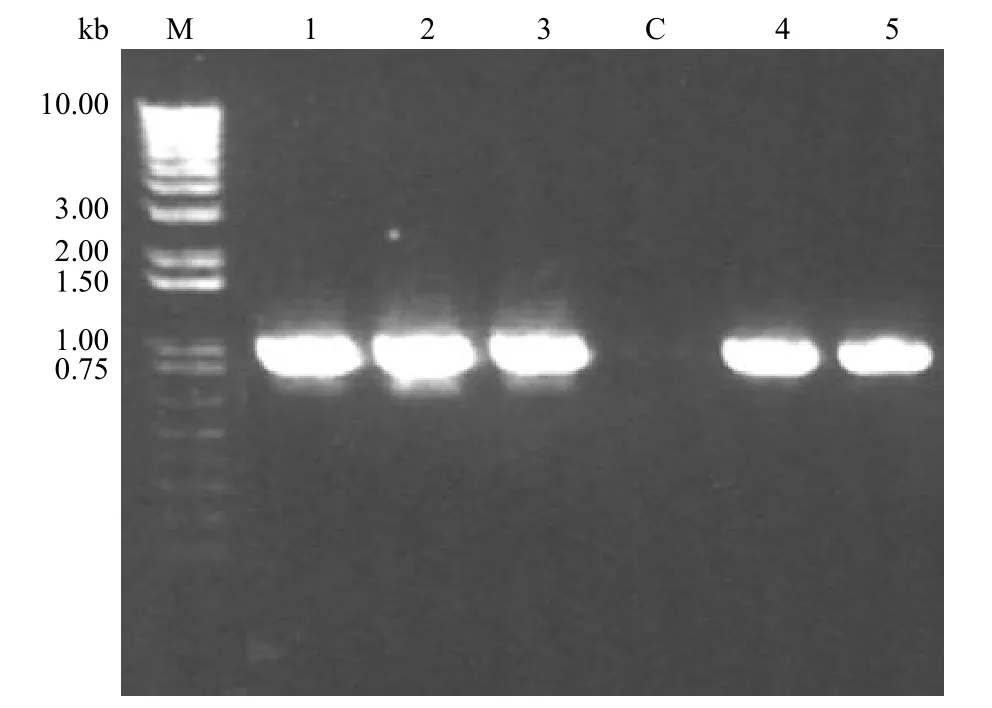
图4 5 个 CYP3a11 基因敲除 E14 ES 克隆的 PCR 检测结果Figure 4 PCR result of five single E14 ES cell colonies with CYP3a11 gene knockouted
2.3 CYP 3a11 基因敲除的 E14 ES 细胞株的 PCR检测
理论上,针对 P3,P4 的扩增产物将横跨 SA和 PGK neo 基因,如果 CYP3a11 基因已经被敲除,所得 PCR 产物应为830 bp 的片段。97 个单克隆分批进行 PCR 检测,其中 5 个单克隆的PCR 检测条带比较清晰稳定,产物均为预期大小的片段(图4)。
2.4 CYP 3a11 基因敲除的 E14 ES 细胞株的Southern 检测
5 个 PCR 检测为阳性的 E14 ES 单克隆提取的总 DNA 经HindIII 酶切后将消化产物琼脂糖凝胶电泳、转膜及 Southern 杂交。理论上,酶解产物将包括约 9 kb neo-LA 片段(图1),可被 neo探针特异结合。如图5 所示,Southern blot 结果表明,这 5 个单克隆中都有 neo 基因的整合。结合PCR 检测结果可以证明,在这 5 个克隆中CYP3a11 基因在 LA 和 SA 序列处已发生重组,部分序列已被 PGK neo 基因所取代。

图5 5 个 CYP3a11 敲除 E14 ES 克隆的 Southern 印迹检测结果,所有 DNA 均用 Hind III 消化Figure 5 Southern blot result of five single E14 ES cell colonies with CYP3a11 gene knockouted, all DNA were digested by Hind III
3 讨论
CYP450 酶系被证明与药物、毒物代谢,转化及致癌密切相关,但目前对其机制的解析远未清晰。由于 CYP450 酶系种属间存在差异,各个酶之间代谢谱出现频繁的交叠及代偿,长期以来基于动物实验的药代动力学及药效学研究结果无法直接推及至人,从而导致了许多新药研究的搁浅。近年来通过基因操作技术而建立的 CYP450 基因敲除小鼠整体模型,如:mEH[4]、sEH[5]、CYP1a[6]、CYP1b1[7]、CYP1a2[8]、CYP2e1[9]基因敲除小鼠,以及 CYP 1a1/1a2[10]、CYP 2g1[11]、CYP 4b1[12]、CYP7a1[13]、CYP2d6[14]、CYP3a4[15]、PXR-null/SXR转基因[16]等人源化小鼠等,为解决上述问题提供了便利。
本研究第一部分构建了小鼠 CYP3a11 基因敲除替换型载体 pCR-CYP3a11_KO,为提高中靶率,我们注意了以下几个方面的问题:⑴运用了长链 PCR 的方法。为避免碱基发生突变,我们采用了 Stratagene 公司生产的目前保真度最高的PfuUltra™ 酶进行基因敲除载体同源臂的扩增,采用热启动以最大限度降低产物背景,将循环次数控制在 30 次。同时对重组片段进行序列分析和酶切鉴定,保证了载体插入同源臂的正确性;⑵选择 CYP3a11 基因的上游 3 号外显子为敲除序列。CYP3a11 蛋白全序列共 504 个氨基酸,在基因 3 号外显子区域插入 PGK neo 基因,可使CYP3a11 基因仅表达上游 95 个氨基酸的短肽。这样可以保证上游同源臂的正确锚定,又能破坏下游重要结构域的表达;⑶尽量增加同源臂的长度。替换型载体同源重组的发生率与同源臂的总长度呈正相关,pCR-CYP3a11_KO 中同源臂总长度约为8 kb(其中 5’-同源臂 0.65 kb,3’-同源臂 7.3 kb),一方面有利于提高重组效率,另一方面短臂有利于用 PCR 法进行初步的筛选鉴定;⑷在载体中选择正负筛选双标记的 neo 和 tk 基因。neo 基因位于长短同源臂之间可以保证载体与基因组间同源重组时 neo 基因能整合到基因组中,而 tk 基因在长臂的外侧,这样可以保证在有效的同源重组发生时tk 基因并不整合至基因组上,防止 tk 表达代谢Ganc 毒杀细胞,从而提高了中靶率。本研究第二步通过电转染该载体进入 ES 细胞成功筛选获得了 5 个基因敲除的 E14 ES 细胞阳性克隆,也证明了该基因敲除载体的构建是正确的。但需要指出的是,本研究所得 5 个阳性 E14 ES 细胞克隆仍有可能由于转染和筛选过程中 tk 基因受损或整合的染色体位置抑制其表达而出现假阳性,因此仍需进一步进行验证。
总之,本研究得到了 CYP3a11 基因替换型打靶载体 pCR-CYP3a11_KO,并通过电转染的方法将其转入 129 系小鼠的 E14 ES 细胞,通过筛选和鉴定获得了 CYP3a11 基因敲除的 E14 ES 细胞阳性克隆,为下一步构建 CYP3a11-null 小鼠奠定基础。
[1]Yanagimoto T, Itoh S, Sawada M, et al.Mouse cytochrome P450(Cyp3a11): predominant expression in liver and capacity to activate aflatoxin B1.Arch Biochem Biophys, 1997, 340(2):215-218.
[2]Nelson DR.Cytochrome P450 and the individuality of species.Arch Biochem Biophys, 1999, 369(1):1-10.
[3]Gonzalez FJ.CYP3A4 and pregnane X receptor humanized mice.J Biochem Mol Toxicol, 2007, 21(4):158-162.
[4]Shin EJ, Bing G, Chae JS, et al.Role of microsomal epoxide hydrolase in methamphetamine-induced drug dependence in mice.J Neurosci Res, 2009, 87(16):3679-3686.
[5]EnayetAllah AE, Luria A, Luo B, et al.Opposite regulation of cholesterol levels by the phosphatase and hydrolase domains of soluble epoxide hydrolase.J Biol Chem, 2008, 283(52):36592-36598.
[6]Nukaya M, Moran S, Bradfield CA.The role of the dioxin-responsive element cluster between the Cyp1a1 and Cyp1a2 loci in aryl hydrocarbon receptor biology.Proc Natl Acad Sci U S A, 2009,106(12):4923-4928.
[7]Tang Y, Scheef EA, Gurel Z, et al.CYP1B1 and endothelial nitric oxide synthase combine to sustain proangiogenic functions of endothelial cells under hyperoxic stress.Am J Physiol Cell Physiol,2010, 298(3):C665-C678.
[8]Hakk H, Diliberto JJ, Birnbaum LS.The effect of dose on 2,3,7,8-TCDD tissue distribution, metabolism and elimination in CYP1A2 (-/-) knockout and C57BL/6N parental strains of mice.Toxicol Appl Pharmacol, 2009, 241(1):119-126.
[9]El Hadri L, Chanas B, Ghanayem BI.Comparative metabolism of methacrylonitrile and acrylonitrile to cyanide using cytochrome P4502E1 and microsomal epoxide hydrolase-null mice.Toxicol Appl Pharmacol, 2005, 205(2):116-125.
[10]Uno S, Endo K, Ishida Y, et al.CYP1A1 and CYP1A2 expression:comparing 'humanized' mouse lines and wild-type mice; comparing human and mouse hepatoma-derived cell lines.Toxicol Appl Pharmacol, 2009, 237(1):119-126.
[11]Zhuo X, Schwob JE, Swiatek PJ, et al.Mouse cyp2g1 gene: promoter structure and tissue-specific expression of a cyp2g1-lacz fusion gene in transgenic mice.Arch Biochem Biophys, 2001, 391(1):127-136.
[12]Imaoka S, Hayashi K, Hiroi T, et al.A transgenic mouse expressing human CYP4B1 in the liver.Biochem Biophys Res Commun, 2001,284(3):757-762.
[13]Agellon LB, Drover VA, Cheema SK, et al.Dietary cholesterol fails to stimulate the human cholesterol 7alpha-hydroxylase gene (CYP7A1)in transgenic mice.J Biol Chem, 2002, 277(23):20131-20134.
[14]Miksys SL, Cheung C, Gonzalez FJ, et al.Human CYP2D6 and mouse CYP2Ds: organ distribution in a humanized mouse model.Drug Metab Dispos, 2005, 33(10):1495-1502.
[15]Granvil CP, Yu AM, Elizondo G, et al.Expression of the human CYP3A4 gene in the small intestine of transgenic mice: in vitro metabolism and pharmacokinetics of midazolam.Drug Metab Dispos,2003, 31(5):548-558.
[16]Xie W, Barwick JL, Downes M, et al.Humanized xenobiotic response in mice expressing nuclear receptor SXR.Nature, 2000, 406(6794):435-439.
猜你喜欢
环球时报(2022-09-20)2022-09-20
汉字汉语研究(2021年2期)2021-08-30
昆明医科大学学报(2021年2期)2021-03-29
今日农业(2020年24期)2020-12-15
中国现代医药杂志(2020年10期)2020-12-14
汉字汉语研究(2019年2期)2019-08-27
新高考·英语进阶(高二高三)(2018年8期)2018-01-15
河北书画研究(2016年3期)2016-04-28
小资CHIC!ELEGANCE(2015年14期)2015-09-23
小资CHIC!ELEGANCE(2015年15期)2015-09-01
