
高渗氯化钠羟乙基淀粉 40注射液对内毒素性急性肺损伤大鼠肿瘤坏死因子-α表达的影响
2010-04-27 10:20宁夏医科大学附属医院麻醉科银川750004高玉华孟尽海张冬梅
陕西医学杂志 2010年9期
宁夏医科大学附属医院麻醉科(银川 750004) 马 涛 高玉华 孟尽海 张冬梅
急性肺损伤 (Acute lung injury,ALI)是临床常见的危重病症,发病机制错综复杂,病死率高达 50%~ 70%[1~3],而全身炎症反应 (Systemic inflammatory response,SIR)是导致 ALI的根本原因[4,5]。机体受到严重创伤、感染、休克和其他致病因素间接或直接刺激后,引起 SIR,此过程涉及众多细胞因子和炎性介质,形成复杂的炎性反应网络。大量研究证实,以多形核中性粒细胞(Polymorpho-nuclear neutrophils,PMN)为主的炎性细胞与多种细胞因子在 ALI的发病过程中起着重要作用,如何调控炎性细胞因子的过度释放,使细胞因子网络恢复平衡状态已成为研究热点。
高渗氯化钠羟乙基淀粉 40注射液作为新型的羟乙基淀粉,因其取代级低、C2/C6比率高且含有 4.2%的氯化钠及 7.6% 的羟乙基淀粉,已较广泛应用于创伤或失血性休克复苏中。由于 HSH40在休克治疗中表现出的特性与 ALI/ARDS相符,故本研究通过复制大鼠内毒素性急性肺损伤模型,将 HSH40用于 ALI的早期液体治疗,观察血气、肺湿 /干重(W/D)比值和肺组织髓过氧化物酶(MPO)活性,各时相血清中 TNF-α表达的变化及肺组织的病理改变,探讨 HSH40对内毒素性急性肺损伤保护作用及可能机制。
材料与方法
1 实验动物 健康成年雄性 SD大鼠 54只,体重 250~ 300g,由宁夏医科大学动物实验中心提供。实验前分笼饲养 1周,自由摄取食水。实验前禁食 12h,禁水 2h。
2 药物与试剂 LPS(E coli O55∶B5)采购于美国 Sigma公司,HSH40由上海长征富民金山制药有限公司提供(批号:H20041554),MPO测试盒购自南京建成生物工程研究所,TNF-αELISA试剂盒购于武汉博士德生物工程有限公司。
3 动物模型与分组 将 54只大鼠随机分为 3组,每组 18只:空白对照组(C组 )、急性肺损伤组 (L组)和 HSH40组 (H组),每组根据时间点分为 3个亚组(亚组 n=6),即 LPS致伤后 0.5h、2h和 4h。各组动物用 3%的戊巴比妥钠 30mg/kg腹腔注射麻醉,L组、H组经股静脉注入 LPS 5 mg/kg致伤,复制急性肺损伤模型[6],C组经股静脉注入等体积的 0.9%NS。致伤1min后 L组从股静脉持续输注 0.9%生理盐水(0.9%NS)5 ml/kg,H组持续输注 HSH405ml/kg;C组用等量 0.9%NS作空白对照,10min输完[7]。
4 观察指标
4.1 血气分析 于致伤后 4h经股动脉采集动脉血 0.5ml,即刻在 i◦ STAT便携式手持血液分析仪上测定 pH、PO2、PCO2值,记录血气分析结果。
4.2 肺 W/D测定 取出右肺下叶,滤纸吸干表面水分,置于干燥称量纸上称得湿重(W);置 65℃恒温箱,烘烤 48h至恒重后称量干重(D),并计算 W/D。
4.3 测定肺组织 M PO活性 右肺剩余组织在冷生理盐水中漂洗,除去血液,滤纸拭干,精确称重后,按重量体积比 1∶19加入 0.86%的冷生理盐水冰浴中制备 5%组织匀浆(匀浆时间 10s/次,间隙 30s,连续 3次)。按照试剂盒说明书步骤,在 460nm处通过比色法测定 MPO活性(单位:活力单位,U/g组织湿片)。
4.4 血清中 TNF-α的测定 分别在大鼠致伤后0.5h、2h和 4h时间点,经右侧股动脉采血 2ml,室温下静置 20min,离心 (4℃,3000r/min,10min)取上清,装入无菌 EP管后,置-80℃低温冰箱中保存。标本收集完整后,采用 ELISA法按照试剂盒说明书步骤,应用全自动酶标定量测定仪进行检测,根据样品的吸光度(OD值)可在绘制出的标准曲线上查出其浓度。
4.5 肺组织病理形态学观察及 ALI病理评分4h组动物处死后,取右肺组织(1cm×1cm×0.5cm)放入 10%中性甲醛溶液固定 48h,常规脱水、透明、浸蜡、石蜡包埋、切片 (5 μ m)、脱蜡、脱水、苏木素伊红(Hematoxylineosin,HE)染色、脱水、封片后光镜下观察肺组织病理形态变化。按照 Mikawa等[8]的方法从四项指标进行肺损伤评分:①肺泡充血;②出血;③间隙或血管壁中性粒细胞浸润或聚集;④肺泡间隔增厚或透明膜形成,根据每项指标病变轻重进行 0~ 4分半定量分析(0:无或极轻微损伤;1:轻度损伤;2:中度损伤;3:重度损伤;4:极重度损伤),累加各项评分的总分作为 ALI的病理评分。用 Olympus BX-51显微镜对病理切片摄片。
5 统计学处理 应用 SPSS11.5统计软件进行分析处理。正态分布的计量资料以均数±标准差(±s)表示,组内各时间点比较采用单因素方差分析,同一时间点组间比较采用配对t检验,以 P<0.05为有显著性差异,P<0.01为有极显著性差异。
结 果
1 动脉血气分析 见表 1。LPS致伤后,大鼠出现低氧血症,呼吸性碱中毒。H组较 C组无显著性差异(P> 0.05),H组和 C组 pH、PO2明显高于 L组(P<0.01),而 PCO2明显低于 L组 (P<0.01)。 H组的PO2、PCO2及 pH值较 L组有明显改善(P<0.01)。
2 肺 W/D、M PO活性及肺损伤病理评分 见表2。 L组肺 W/D、M PO活性及肺损伤病理评分显著高于 C组(P<0.01),H组上述各项指标均明显下降,与L组相比有显著性差异(P<0.01)。
表1 各组大鼠动脉血气的变化 (±s)

表1 各组大鼠动脉血气的变化 (±s)
注:*与 C组比较,P<0.01;#与 L组比较,P<0.01
组 别 n pH PO2(mmHg) PCO2(mmHg)C 组 18 7.39± 0.02 91.6± 2.41 34.68± 2.42 L 组 18 7.28± 0.02* 56.3± 4.73* 45.66± 1.02*H组 18 7.38± 0.03# 88.7± 2.26# 34.84± 2.66#
表2 肺 MPO、W/D及病理评分结果 (±s)
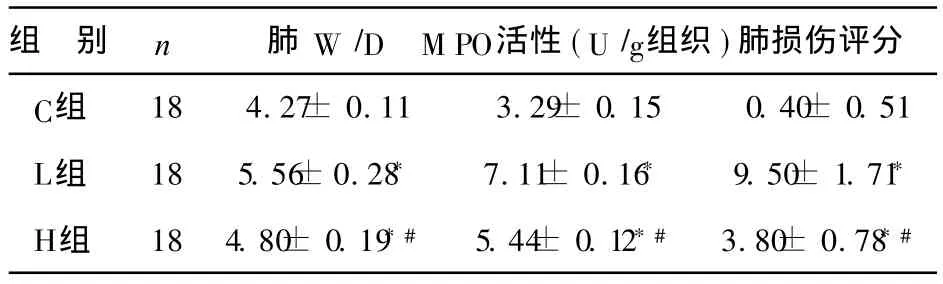
表2 肺 MPO、W/D及病理评分结果 (±s)
注:*与 C组比较,P<0.01;#与 L组比较,P<0.01
组 别 n 肺 W/D M PO活性(U/g组织)肺损伤评分C组 18 4.27± 0.11 3.29± 0.15 0.40± 0.51 L组 18 5.56± 0.28* 7.11± 0.16* 9.50± 1.71*H组 18 4.80± 0.19*# 5.44± 0.12*# 3.80± 0.78*#
3 血清 TNF-α的变化 见表 3。 C组 TNF-α的含量在 3个时间点均无显著性差异(P>0.05);L组 3个时间点两两间均有显著性差异(P<0.01),0.5h表达开始升高,2h达峰值,以后逐渐下降;而 H组 TNF-α的含量与 L组相比明显降低,但仍高于 C组(P<0.01),H组内 0.5h~ 2h和 2h~4h有显著性差异(P<0.01),0.5h~ 4h无显著性差异 (P> 0.05)。
表3 各亚组大鼠血清中 TNF-α含量的变化(±s,pg/ml)

表3 各亚组大鼠血清中 TNF-α含量的变化(±s,pg/ml)
注:组内比较:*与 0.5h比较,P<0.01;#与 2h比较,P<0.01;组间同一时间点比较:▲与 C组比较,P<0.01;★与 H组比较,P<0.01
C组 6 49.23± 2.69 49.26± 2.55 47.61± 2.12 L组 6 201.90± 9.05▲ 346.02± 13.25★*180.83± 11.32▲*#H组 6 158.69± 7.91▲★259.85± 17.99▲★*158.54± 5.28▲★#
4 肺组织病理形态学变化 见图 1~ 3。光镜下,C组肺组织结构完整,肺泡结构清晰,肺泡腔完整,壁光滑,肺泡间隔均匀一致,可见少量白细胞,肺泡腔中无渗出液或渗出的白细胞;L组肺泡隔明显增宽,肺泡壁完整结构破坏,肺泡壁断裂,部分肺泡内见出血及微血栓形成,大量白细胞渗出、聚集,肺泡腔中可见渗出液、红细胞、多形核中性粒细胞、巨噬细胞,部分肺泡腔萎陷不张 ;H组肺泡壁有轻度水肿,肺间隔略有增厚,但出血和炎性细胞浸润情况较 L组明显减轻,肺组织结构基本完整。

图1 C组肺组织形态学改变(HE×100)

图2 L组肺组织形态学改变(HE×100)

图3 H组肺组织形态学改变(HE×100)
讨 论
革兰阴性杆菌是临床上引起感染的常见微生物,其主要致病物质是内毒素(ET),而 LPS是 ET的主要成分,参与构成革兰阴性菌的细胞壁,是强诱导剂。LPS趋化、激活中性粒细胞(PMN)在肺间质和肺泡腔内大量聚集、释放炎性细胞因子、氧自由基等,损伤肺泡-毛细血管膜,使之通透性升高,形成了肺损伤的基本病理改变,与 ALI的发生密切相关[9,10]。本实验模型组(L组)在注射 LPS 4h后,PO2、pH明显下降,PCO2升高,肺 W/D显著增加,肺脏出现了 ALI的病理学改变,肺损伤评分(9.50±1.71分 )明显升高,说明模型复制成功。
TNF-a是创伤或感染后机体最早产生的多功能细胞因子之一,主要由单核细胞和巨噬细胞产生。Ayata等[11]的研究表明 TNF-a可能是炎性介质链锁反应中最早的启动因子,对肺有强烈毒性可以通过多种机制引起肺损伤。它可以促使 PMN趋化、聚集、黏附于血管内皮细胞并脱颗粒释放溶酶体,产生氧自由基等,还能促使单核细胞和巨噬细胞的分化,并使巨噬细胞和内皮细胞释放 IL-1、IL-6等其他细胞因子而形成级联反应。
本实验结果显示大鼠血清中 TNF-a于注射 LPS后 0.5h开始升高,2h达峰值,以后逐渐降低,经过HSH40治疗后,TNF-a含量明显下降,以 2h最为显著。表明在 ALI早期,TNF-a的合成和分泌亢进,这种变化导致致炎 /抗炎失衡,通过下列途径引起 ALI的发生[12,13]:①增加肺血管通透性,引起肺水肿、低血压;②上调 PMN、肺血管内皮细胞对补体片段 CR3以及细胞间黏附分子(ICAM-1)、P选择素等黏附分子的表达,促进 PMN与内皮细胞的黏附及迁移;③延迟 PMN凋亡,增加 PMN的细胞毒性及内皮细胞对 PMN介导的杀伤效应的敏感性。而 HSH40在 ALI早期可以抑制TNF-α的合成与表达,使细胞因子网络恢复平衡状态,从而减轻肺损伤的发生,发挥肺脏保护作用。
MPO是存在于 PMN嗜天青颗粒中特有的酶 ,心脏、肾和皮肤等疾病的研究证实,M PO活性是评价PMN在组织中扣押浸润程度的可靠指标[14]。实验显示,MPO活性在 C组无明显改变,L组在 4h显著升高,而 H组虽高于 C组,但明显低于 L组。说明 HSH40可以抑制 PMN的呼吸爆发,减轻氧自由基等活性物质对肺的损伤。
HSH40是高渗晶体 /胶体混合液,具有比生理盐水或其他等渗输液高 4~ 5倍的渗透压浓度(渗透压为1400mOsm/L)。通过高渗氯化钠产生的渗透压梯度使水分从细胞内、组织间隙转移至血管内,以自体输液的形式快速主动扩充血容量,直接改善微循环;羟乙基淀粉则可以增加胶体渗透压,稳定和维持有效循环血容量,从而减轻 ALI时的毛细血管渗漏和肺水肿。目前认为高渗晶 /胶液对肺损伤的影响和可能机制有以下几个方可面:①抑制炎性细胞间和细胞内的信号传导;②减少中性粒细胞在肺内的扣押和减轻菌群移位,减少内毒素血症的发生,消除由此介导的 PMN呼吸爆发;③打破致炎效应和抗炎效应之间的平衡,上调抗炎效应;④减少细胞因子和黏附分子的表达。
综上所述,在内毒素性 ALI时,炎性因子 TNF-a在肺组织中大量产生和表达,PMN黏附、聚集、扣押,肺微血管通透性增高,产生过度炎症反应。 HSH40通过阻断 TNF-a的合成与释放,降低肺微血管通透性,减少白细胞浸润和减轻炎症反应,从而对 ALI产生保护作用。
[1] Stapleton RD,Wang BM,Hudson LD,et al.Causes and timing of death in patients with ARDS[J].Chest,2005,128(2):479-481.
[2] Luh SP,Chiang CH.Acute lung injury/acute respiratory distress syndrome(ALI/ARDS):the mechanism,present strategies and future perspectives of therapies[J].Zhejiang Univ Sci B,2007,8(1):60-69.
[3] 邱海波.规范和提高我国急性呼吸窘迫综合征的诊断和治疗 [J].中国危重病急救医学,2006,18(12):705.
[4] 钱桂生.急性肺损伤和急性呼吸窘迫综合征研究现状与展望.解放军医学杂志,2003,28(2):97.
[5] Bhatia M,Moochhala S.Roleof inflammatory mediators in the pathophysiologyofacuterespiratorydistress syndrome.J Pathol,2004,202(2):145.
[6] YamasawaH,Ishii Y,Kitamura S.Cytokine-induced neutrophil chemoattractant in a rat model of lipopolysaccharide-induced acute lung injury.Inflammation,1999,23(3):263-274.
[7] Russell DH,Barreto JC,Klemm K,et al.Hemorrhagic shock increases gut macromolecular permeability in the rat[J].Shock,1995,4(1):50.
[8] Mikawa K,Nishina K,Takao Y,et al.ONO-1714,a nitric oxide synthase inhibitor,attenuates endotoxin-induced acute lung in rabbits.Anestth Analg,2003,97(6):1751-1755.
[9] Ulitck T,Watson Y,Songmei K,et al.The intratrachcal dministration of lipopolysacchharide and cytokines:characterization of LPS-induced IL-1,and TN F-inflammatory infiltrate.Am J Pathol,1991,138(6):1485-1496.
[10] Jorg R,Klaus L.Bench-to-bedside review:acute respiratory distress syndrome-how neutrophils migrate into the lung.Crit Care,2004,8(6):453-461.
[11] Ayata A.Perrin MM,M eldnum DR,et al.Hemorrhage induced an increase in serum TNF which is not associated with elevated levels of endotoxin.Cytokine,1990,16(2):170.
[12] Goodman RB,Pugin J,Lee JS,et al.Cytokine-mediated inflammation in acute lung injury[J].Cytokine Growth Factor Rev,2003,14(6):523-535.
[13] Shimabukuro DW,Sawa T,Gropper M A.Injury and repair in lung and Airways[J].Crit Care Med,2003,31(Supp1):524-531.
[14] Rizoli SB,Rotstein OD,Parodo J,et al.Hypertonic inhibition of exocytosis inneutrophils:central role for osmotic actin skeleton remodeling.Am J Physiol Cell Physiol,2000,279(3):619-633.
猜你喜欢
现代临床医学(2021年4期)2021-07-31
猪业科学(2021年3期)2021-05-21
中华养生保健(2020年9期)2021-01-18
心肺血管病杂志(2020年5期)2021-01-14
国际呼吸杂志(2019年8期)2019-04-29
国际呼吸杂志(2019年8期)2019-04-29
老年医学与保健(2017年6期)2017-02-06
中外医疗(2016年15期)2016-12-01
湖南中医药大学学报(2016年1期)2016-12-01
中华老年多器官疾病杂志(2016年9期)2016-04-28
