
水化膜对石英表面硫化作用的DFT研究
2024-12-08 00:00:00李建新
中国新技术新产品 2024年3期
关键词:硫化




摘 要:在许多地质检测和工业生产的过程中,硫化是一种常用的改善氧化矿浮选的预处理工艺。并不是所有的氧化矿物都可以硫化,因此石英表面的硫化十分重要。然而,该过程的详细机理目前仍不清楚,尤其是在存在水化膜的情况下。因此,本文使用密度泛函理论(DFT)来研究水化膜对石英表面硫化作用的影响。计算结果表明,水化膜对石英能否硫化起决定性作用,石英中的Si-O键比Si-S键强烈,导致石英表面水化膜对硫化氢(HS)有强大障碍,石英的硫化难以进行。为找出其内部机理,本文进一步分析了重叠布居、投影态密度(PDOS)和电子构型,并建立了它们与硫化行为的关系,研究其对硫化产物的性质和稳定性有何影响。
关键词:DFT计算;硫化;水化膜;氧化物矿石
中图分类号:TD 87 " " " " " " " 文献标志码:A
浮选是一种矿物加工方法,它利用矿物表面物理和化学性质的差异,在冶炼前对矿物进行分选。硫化-浮选是许多氧化物矿石选矿的常用方法,该方法使用硫化试剂对矿物进行预处理,然后加入捕收剂进行浮选[1]。硫化是指预处理的过程。然而,并非所有氧化矿物都适合采用硫化-浮选法进行选矿,因为有些氧化物矿石能够硫化,有些则不能。石英就是一个典型的不能够硫化的氧化物矿石,表面水化膜在石英硫化过程中的作用也很模糊。仅依靠试验研究还不能深刻理解石英表面水化膜的形成对其硫化的具体作用,理论研究是必要的。因此,本文采用密度泛函理论(DFT)进行计算模拟。计算结果揭示了水化膜对石英硫化作用的影响,并通过分析重叠布居、投影态密度(PDOS)和电子构型,研究了原子水平上的更多信息及其潜在机理。
1 计算方法与理论模型
1.1 计算方法
本文采用第一性原理计算方法,直接从原子核与电子的角度出发对反应体系进行理论模拟。微观尺度下粒子的行为受量子力学约束,需要使用薛定谔方程进行描述。然而直接求解多原子体系的薛定谔方程计算量巨大,难以应用于实际体系中。基于密度泛函理论框架求解方程能够显著降低计算量,同时也能避免对计算精度的影响,是目前应用最广的计算方法。因此,本文使用VASP(Vienna Abinitio Simulation Package)软件并基于密度泛函理论框架求解体系的薛定谔方程[2-4],使用广义梯度近似(GGA)的能量近似泛函(Perdue-Burke-Ernzerhof,PBE)版本描述交换-关联相互作用[5],并使用高精度投影缀加波(PAW)方法对核-价电子相互作用进行建模[6-7]。
1.2 理论模型
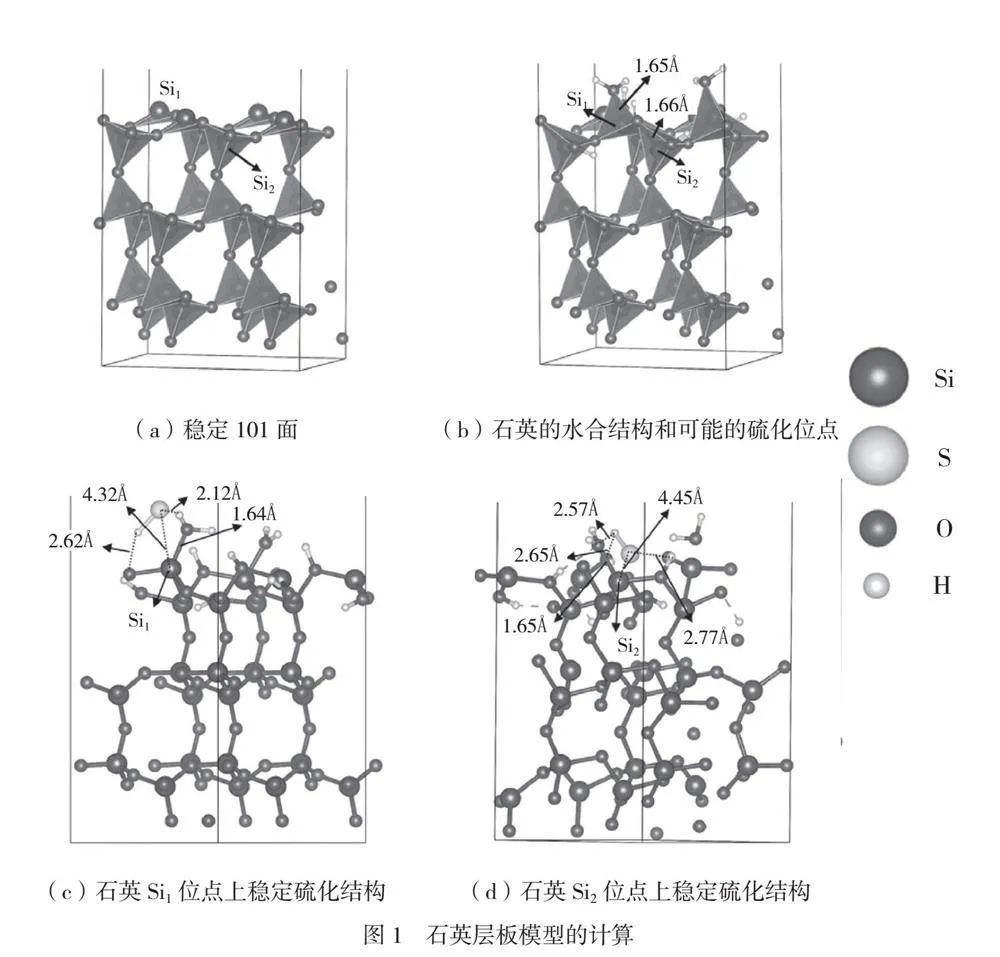
为了准确预测体相性质并进行表面特性计算,在具体参数上,本文使用截断能550eV、0.12Å的有限网格间距和密度为12×12×12的Monkhorst-Pack K点网格。能量收敛精度定位1×10-5eV/atom,单原子所受最大压力为0.01eV/Å。石英层板模型的计算如图1所示。在图1(a)中,本文选取了热力学最稳定的(101)晶面进行研究,在垂直方向选取了包括3层O-Si-O叠层结构的2×2的超晶胞。为了避免相邻重复单元间发生相互作用,在垂直方向添加了15Å的真空层区域。在石英表面模型中,顶部2个原子层上的原子可以移动,而其他原子层不能移动,对计算能量差别的影响较小。Si是石英可能的硫化位点,为了研究Si在水化和硫化中的作用,石英表面模型上的Si位有3-配位Si(Si1)和4-配位Si(Si2)2种。在具体参数上,使用了截断能500eV,k点采取伽马点3×3×1的网格点。能量收敛精度定位1×10-6eV/atom,单原子所受最大压力为0.02eV/Å。HS和H2O的平衡结构采用伽马点在(15×15×15)Å3的盒子进行优化。
计算H2O/HS吸附到石英表面的程度时,通常用吸附能Eads来表示,其计算过程如公式(1)所示。
Eads=Esur+Eadsorbate–Esur+adsorbate " "(1)
式中:Eads(kJ/mol)为吸附能;Esur(kJ/mol)和Esur+adsorbate(kJ/mol)分别为吸附H2O/HS前、后石英表面优化后的总能;Eadsorbate为吸附分子H2O/HS在盒子中的总能。
对HS和H2O分子结构进行优化,单独将其放在15×15×15Å3的盒子中得到其平衡结构,使用了截断能450eV、3×3×1的伽马网格点,能量收敛精度定位1×10-7eV/atom。
2 结果与讨论
2.1 水合结构
石英的稳定水合结构如图1(b)所示。石英的平均表面水化能为-598.973kJ/mol,说明石英的表面水化在热力学上是稳定的。在石英表面的水合过程中,水分子会解离成OH-和H+,分别与表面位点成键形成一层水化膜。Si1和Si2位点上的Si-O键在石英水化结构中形成,其键长分别为1.65Å和1.66Å,明显小于Si-O的共价键半径1.84Å。石英水合结构中Si-O的重叠群体值见表1。从表1可以看出,石英水合结构中Si-O的相应重叠群体值均为正值,表明这种矿物发生了水合作用;Si1-O和Si2-O的对应重叠群体值分别为0.54和0.56,说明石英水合结构形成了稳定的Si-O键。因此,Si-O键可能难以被破坏。
2.2 硫化结构
HS分子在石英硫化结构中的吸附能见表2。HS分子在Si1位点的吸附能为-28.87kJ/mol,而在Si2位点的吸附能为-22.62kJ/mol。这些吸附能值显然不是硫化反应引起的,因为它们均接近零,表明这些相互作用更可能是物理吸附而非化学吸附。通过观察图1(c)和图1(d)中已优化的石英硫化结构,可以进一步解释该现象。在石英的硫化结构中,Si1-S键(键长为4.32Å)和Si2-S键(键长为4.45Å)长度大于2个原子共价半径,未形成Si-S键,表明硫化反应没有发生。此外,Si1-O键(键长为1.64Å)和Si2-O键(键长为1.65Å)也没有断裂,其键长明显小于其共价键半径(1.84Å),进一步表明没有化学反应发生,但Si1和Si2位点的负吸附能较小,原因是石英硫化结构中形成了H键。进一步观察如图1(c)所示的石英硫化结构,可以发现Si1位点上形成了O-H键(键长为2.62Å)和S-H键(键长为2.12Å)。如图1(d)所示,Si2位点的硫化结构中形成了2个O-H键(键长分别为2.57Å和2.65Å)和一个S-H键(键长为2.77Å)。因此,不是硫化反应,而是硫化结构中形成的H键被认为是导致Si1和Si2位点上出现近零吸附能的主要原因,这进一步支持了石英的硫化未发生的结论。
石英硫化结构中的Si1-O、Si1-S、Si2-O和Si2-S的重叠布居值见表3,Si1-S和Si2-S的重叠布居值为负值,说明石英表面没有发生硫化。同时,Si1-O和Si2-O的正重叠布居值则说明石英表面水合膜上的Si-O键未因HS的作用而被破坏。这与上述分析结果一致,石英表面水合膜可抑制HS的作用。
2.3 态密度(DOS)
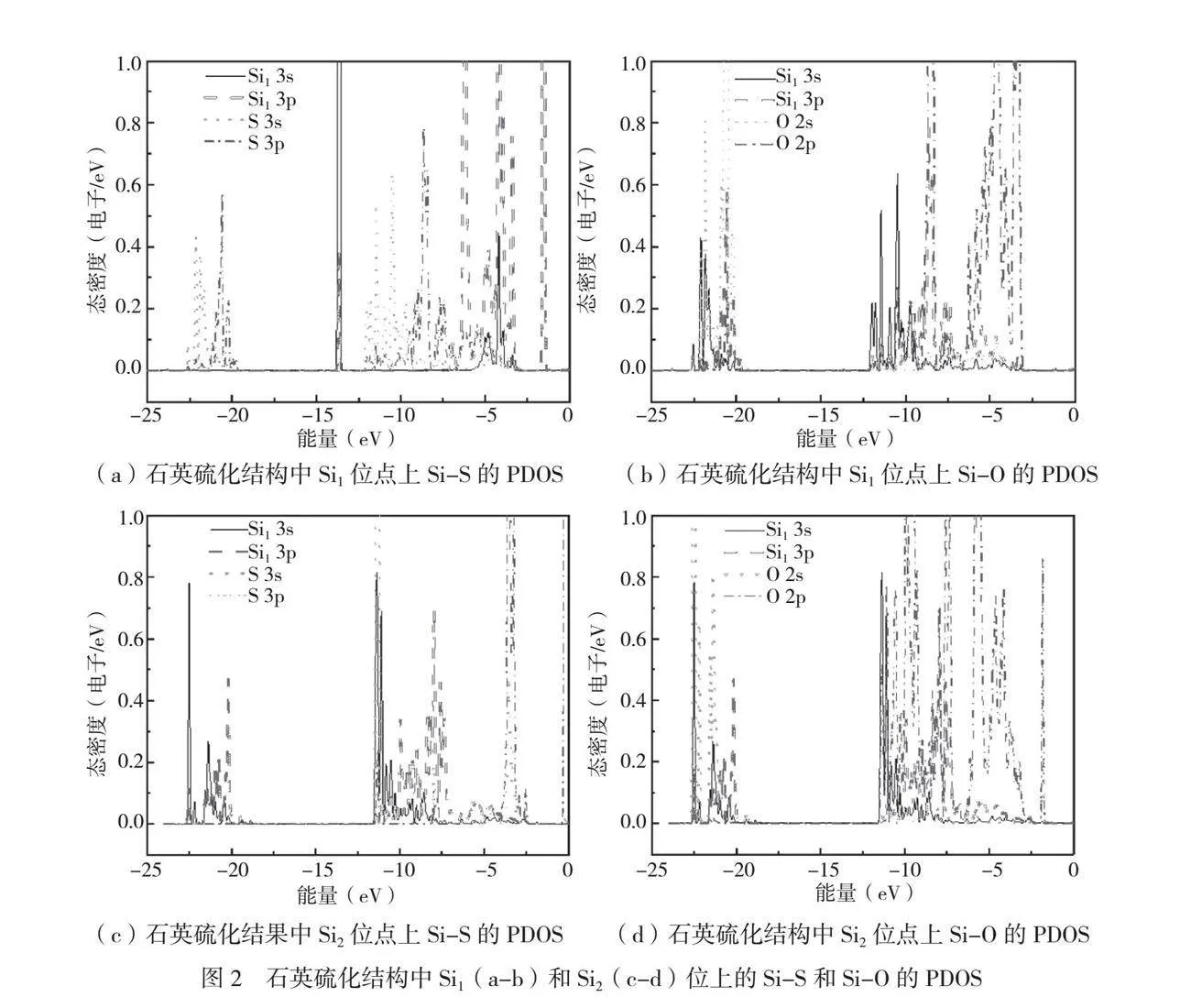
为了探究电子水平的作用机理,石英硫化结构中Si1和Si2位置的硅-硫(Si-S)键的PDOS如图2所示。从图2(a)中可以发现,在-25.0eV~-12.5eV和-10.0eV~-5.0eV的能量范围内,没有观察到S 3s、S 3p轨道和Si1 3s、Si1 3p轨道的杂化峰,表明Si1和S间不存在成键。类似地,从图2(c)中可以发现,在-25.0eV~-12.5eV和-7.5eV~-2.5eV的能量范围内,也未观察到S 3s、S 3p轨道和Si2 3s、Si2 3p轨道的杂化峰,说明Si2和S间也无成键。进一步分析Si1和Si2位点上的石英硫化结构中前、后S的PDOS,其变化非常小。因此,分析Si1和Si2位点上的Si-S键,可以进一步确认石英晶体不会发生硫化反应,这些结果表明石英晶体在被表面水化后没有硫化潜力。
石英硫化结构中Si1和Si2位置上Si-O键的PDOS如图2(b)和图2(d)所示。从图2(b)中可以发现,在-22.51eV~20.32eV观察到O的2s轨道与Si1的3p轨道有1个轨道杂化峰,同时在-8.10eV和-6.99eV位置观察到O的2p轨道与Si1的3p轨道的轨道杂化峰,证实了石英表面水化膜上的Si1-O键没有被破坏。类似地,在图2(d)中,O的2s轨道与Si2的3p轨道在-23.11eV~-20.01eV出现了s-p轨道杂化。同时在-10.13eV~-6.08eV,O的2p轨道与Si2的3p轨道也出现了p-p轨道杂化,表明石英表面水合膜上的Si2-O键同样没有被破坏。通过分析Si1-O和Si2-O键的PDOS,进一步确认了石英晶体表面的水合膜的稳定性。这些观察结果对理解石英表面、水分子的相互作用和水化膜的特性具有重要意义。
3 结论
研究结果表明,石英并没有发生硫化反应,其接近正值的吸附能主要由H键相互作用引起。此外,矿物表面的水合薄膜对其硫化性能有一定影响。因此,本文进一步讨论了其基本机理。原子间距和COHP分析结果表明,一方面,Si-S键的长度﹥其共价半径(2.13Å)且不存在Si-S键的重叠布居值,进一步证明了石英没有发生硫化反应。另一方面,Si-O键的长度小于其共价半径(1.84Å),并且Si-O键的正重叠布居值表明石英表面水合膜上的Si-O键未被破坏,即石英表面的水合膜有效地阻止了HS进一步的相互作用。上述分析可以从配位化学中得到解释,H2O中没有空的π轨道,而HS空的π轨道能量过高,反应性非常弱,因此,它们倾向于通过σ键而不是π反键与金属离子相互作用,它们都是电子给予者。然而,由于硫的电负性(2.58)﹤氧的电负性(3.44),因此HS比H2O更容易提供电子。石英表面的Si是硬酸,根据硬软酸碱理论(Hard-Soft-Acid-Base,HSAB),它更倾向于与硬碱H2O结合,而不是与软碱HS结合,因此石英表面的水合薄膜有效地抑制了HS进一步的相互作用。
参考文献
[1]闵凡飞,任豹,陈军,等.基于水化膜弱化促进煤泥脱水机理及试验研究[J].煤炭学报,2020,45(1):368-376.
[2]邱廷省,黄开国.硫化钠对高冰镍中六方硫镍矿的抑制行为研究[J].有色金属,1997(4):18-21.
[3]闵凡飞,任豹,陈军,等.基于水化膜弱化促进煤泥脱水机理及试验研究[J].煤炭学报,2020,45(1):368-376.
[4]刘世强,衣洪源,马武举,等.菱锌矿表面强化硫化技术及机理研究[J].有色金属(选矿部分),2023(2):30-40.
[5]KRESSE G,HAFNER J.Ab initio molecular-dynamics simulation
of the liquid-metal--amorphous-semiconductor transition in
germanium[J].Phys.Rev.B:Condens matter,1994,49(20):14251-14269.
[6]PERDEW J P,BURKE K,ERNZERHOF M.Generalized Gradient Approximation Made Simple[J]Phys.Rev.Lett.,1996,77(18):3865-3868.
[7]KRESSE G,JOUBERT D.From ultrasoft pseudopotentials to the projector augmented-wave method[J].Rev.B:Condens matter,1999,59(3):1758-1775.
猜你喜欢
橡塑技术与装备(2021年11期)2021-06-16 05:43:14
中国特种设备安全(2019年3期)2019-04-22 05:05:38
橡塑技术与装备(2018年1期)2018-12-25 06:34:17
橡胶科技(2018年12期)2018-07-21 02:34:10
山东工业技术(2016年15期)2016-12-01 05:30:43
环境科技(2015年5期)2015-11-08 12:08:58
橡胶科技(2015年6期)2015-07-31 07:04:54
弹性体(2015年1期)2015-06-11 01:30:48
世界橡胶工业(2015年7期)2015-03-04 08:50:30
世界橡胶工业(2014年11期)2014-04-14 03:14:02
