
miR-224-5p 调控PI3K/Akt/FoxO1 轴抑制氧化应激减轻缺氧/复氧诱导的心肌细胞损伤
2024-11-03 00:00:00梁国新唐红悦郭畅张明明
南方医科大学学报 2024年6期
摘要:目的 探究miRNA-224-5p在缺氧/复氧(H/R)诱导心肌细胞损伤中的作用机制。方法 收集160 例急性心肌梗死(AMI)患者和80 例健康体检者(HC)的血浆检测miRNA-224-5p及生化指标。H9c2细胞分为对照组(Control)、H/R组、H/R+miR-224-5pNC组、H/R+miR-224-5p mimics组。噻唑蓝(MTT)检测细胞活力;试剂盒检测丙二醛(MDA)、超氧化物歧化酶2(SOD2)和乳酸脱氢酶(LDH);双荧光素酶报告基因实验验证miR-224-5p与PTEN的靶向关系;生物信息学方法对靶基因的潜在机制进行分析;qRT-PCR 检测miRNA-224-5p mRNA 表达;Western blotting 检测PTEN、Bcl-2、Bax、Cleaved caspase-3、SOD2、p-PI3K/PI3K、p-Akt/Ak 及p-FoxO1/FoxO1 蛋白水平;流式细胞术检测细胞凋亡率。结果 与 HC 组相比,AMI 组的血糖、 C 反应蛋白、CK、CK-MB 和cTnI水平均显著高于对照组(Plt;0.05)。AMI组和H/R组miR-224-5p的表达低于对照组(Plt;0.05);心肌细胞活力呈缺氧/复氧时间依赖性减少;PTEN是miR-224-5p 的靶基因;PI3K/Akt 通路是最显著富集的通路;与Control 组相比,H/R 组SOD2 的活性降低,LDH的活性和MDA的含量均上升,细胞凋亡率上升(Plt;0.05),细胞中p-PI3K、p-Akt、p-FoxO1、SOD2 和Bcl-2蛋白表达水平均降低,PTEN、Bax和Cleaved caspase-3蛋白表达均升高(Plt;0.05);与H/R组比较,H/R+miR-224-5p mimics组SOD2的活性显著上升,LDH、MDA的水平和细胞凋亡率均显著降低(Plt;0.05),细胞中p-PI3K、p-Akt、p-FoxO1、SOD2和Bcl-2的表达均上调,PTEN、Bax和Cleaved caspase3蛋白表达水平均降低(Plt;0.05)。结论 miR-224-5p过表达通过PI3K/Akt/FoxO1轴上调抗氧化基因SOD2的表达,缓解H/R诱导的H9c2细胞的氧化应激,减少细胞凋亡。
关键词:缺氧/复氧;微小RNA;氧化应激;凋亡
缺血性心肌病(ICM)是由冠状动脉粥样硬化性心脏病(冠心病)心肌缺血引起的心肌功能失常,冠心病事件发病率约为35/万~835/万在全球,且呈现逐年上升的趋势[1]。其发病机制包括缺血致心肌梗死、非梗死区域代偿肥大、内皮功能障碍、心肌细胞凋亡等[2]。随着对冠心病研究的日益深入,氧化应激在冠心病发生发展中的意义日益凸显。氧化应激(OS)是当体内氧自由基(OFR)产生过度或清除减少,造成体内活性氧类生成与抗氧化防御之间的平衡紊乱,其在ICM疾病过程中可损害细胞膜,致钙超载、细胞凋亡,产生炎性介质,损伤内皮细胞,损害血小板功能,影响miRNA的表达,继而导致ICM的发生和发展[3]。而急性心肌梗死(AMI)是缺血性心脏病中导致人类死亡的主要疾病之一。
心肌缺血/再灌注损伤(IRI)是心脏手术、心脏移植、心肌梗死和心脏骤停中常见且严重的病理生理现象。IRI的细胞和分子机制非常复杂,包括炎症、内皮功能障碍、钙浓度和pH值的变化,以及线粒体功能障碍和氧化应激[4]。心肌IRI最常发生在急性心肌梗死(AMI)再灌注治疗期间。再灌注治疗可以清除阻塞的冠状动脉并恢复心肌的血流,但可能会导致活性氧(ROS)的产生急剧增加,这会压倒内源性抗氧化系统。过量的ROS不仅会引发氧化应激还会导致心肌细胞凋亡进而加重心肌细胞损伤,这两者都在AMI的发病机制中发挥重要作用[5, 6]。因此,提高机体的抗氧化能力从而消除过多的ROS可能是改善心肌梗死等缺血性心脏病的有效途径。
微小RNA(miRNA)是一种长度约为18~23个核苷酸的内源性非编码微小RNA,负向调节基因表达并调节特定基因的翻译和稳定性。近年来,在I/R损伤的心肌组织中发现了各种miRNA的显著变化,表明miRNA可能是治疗心肌I/R损伤的潜在治疗靶点[7]。LncRNAAK087124/miR-224-5p/PTEN轴通过细胞凋亡和AKT信号通路调节动脉粥样硬化的内皮细胞损伤[8]。miR-224-5p 靶向TXNIP 增强了HP-EV在缓解心肌梗死和再灌注治疗前心肌细胞缺氧状况方面的抗凋亡作用[9]。miR-224-5p/CPEB3 使EGFR/PI3K/AKT 信号失活来抑制食管鳞状细胞癌的发展[10]。PI3K/Akt信号通路的活化在心肌梗死以及心肌缺血再灌注损伤((MI/RI)的心脏保护中起着至关重要的作用,被认为是一条经典的对心脏有利的通路[11]。而叉头转录因子1(FoxO1)是PI3K/Akt信号轴的下游靶点[12]。FoxO1参与包括氧化应激抵抗、代谢调节、细胞增殖和凋亡在内的多种生物学过程[13]。作为调节氧化还原动态平衡的关键转录因子,FoxO1促进位于不同亚细胞室的抗氧化蛋白的表达,如线粒体中的锰超氧化物歧化酶(SOD2)[14]。miR-224-5p与凋亡、炎症和心脑血管病的缺血再灌注损伤等生理病理过程密切相关[15]。然而,miR-224-5p 是否通过调节FoxO1蛋白表达维持心肌细胞内活性氧的水平从而减轻心肌细胞损伤尚未确定。
已证实体外H/R处理的H9c2心肌细胞模型可以复制在体心肌I/R损伤模型[16, 17]。因此在本研究中,我们利用H9c2心肌细胞建立了体外培养的H/R细胞模型,探讨miR-224-5p 通过PI3K/Akt/FoxO1 信号轴抑制氧化应激减轻缺氧/复氧对H9c2心肌细胞的影响。
1 材料和方法
1.1 材料
1.1.1 研究对象
选择2021年12月~2023年2月在河北人民医院心血管内科收治的80 例ST段抬高心肌梗死(STEMI)患者,非ST段抬高心肌梗死(NSTEMI)患者80例。STEMI和NSTEMI患者的诊断基于ACC/AHA/ESC/WHF实践指南[18]。胸痛超过12 h、接受溶栓治疗和患有严重心力衰竭的AMI患者不包括在本研究中。还招募了同期80 名年龄和性别匹配,没有证据表明有心血管或脑血管疾病的健康受试者作为健康对照组(HC)。从MI患者和健康体检者收集5 mL空腹静脉血立即离心以获得纯血浆,随后储存在-80 ℃用于进一步分析。河北省人民医院伦理委员会批准了本研究(伦理审查编号:202104),所有参与者均已知情同意。
1.1.2 材料和试剂
H9c2心肌细胞株(HCM)购于中国科学院上海细胞库;DMEM细胞培养基、Lipofectamine3000、TRizol试剂、miRNA逆转录试剂盒(lnvitrogen);胎牛血清(武汉普诺赛生命科技有限公司);青链霉素双抗溶液、丙二醛(MDA)含量检测试剂盒、超氧化物歧化酶(SOD)活性检测试剂盒和乳酸脱氢酶(LDH)活性检测试剂盒(南京建成生物工程研究所);miR-224-5p 模拟物(miR-224-5p mimics)、模拟物阴性对照(miR-NC)、miR-224-5p抑制物(anti-miR-224-5p)、抑制物阴性对照(anti-miR-NC)以及双荧光素酶报告载体(汉恒生物科技(武汉)有限公司);逆转录酶、2×SYBR Green mastermiX (广州瑞博生物科技有限公司);膜联蛋白-异 硫氰酸荧光素/碘化丙啶(AnneXin V-FlTC/Pl)(汉恒生物科技(武汉)有限公司);兔抗PTEN抗体、兔抗PI3K抗体、兔抗Akt抗体购于(购于Cell Signaling Technology公司);超氧化物歧化酶2抗体(SOD)、磷酸化叉头蛋白家族1抗体(p-FOXO1A (phospho S256))(上海联迈生物工程有限公司);裂解型含半胱氨酸的天冬氨酸蛋白水解酶-3兔单克隆抗体、兔B细胞淋巴瘤-2(Bcl-2)多克隆抗体、兔Bcl相关X蛋白(BaX)单克隆抗体、β-肌动蛋白抗体(β-actin)单克隆抗体、辣根过氧化物酶标记山羊抗兔lgG(上海碧云天生物公司)。
1.2 方法
1.2.1 检测方法
患者入院后的空腹静脉血,离心力调至2180×g,10 min 分离血清及血浆,贝克曼自动生化分析仪对CK、CK-MB、CRP 及血糖进行检测;全自动电化学发光分析仪测定血清中cTnI 的浓度。
1.2.2 H9c2 心肌细胞的培养
H9c2 细胞置于37 ℃、5%CO2的培养箱在含有完全培养基(RPMI 1640+10%胎牛血清+1%青-链霉素溶液)的培养瓶中培养,每3 d换液1 次,当细胞达到85%~90%融合时传代或冻存。参照[19]构建H/R 模型:首先在低葡萄糖DMEM培养基中(1 mg/mL),低氧条件(95% N2 和 5% CO2,37 ℃)置于三气培养箱中孵育12 h。随后,细胞在含10%胎牛血清的DMEM中、在95%空气和5%CO2中37℃孵育不同的时间,不同的时间取决于复氧需要的时间。
1.2.3 细胞转染与分组
取对数期的细胞2×105接种于含有1%FBS 的高糖DMEM培养基(4.5 mg/mL)的24孔板中并孵育24 h。转染miR-NC、miR-224-5p mimics按照Lipofecta mineTM3000 试剂制造商的方案进行转染,48 h 后收集细胞进行H/R 处理,分别记为H/R+miR-NC组(空载体)与H/R+miR-224-5p 组(模拟物序列)。在DMEM 中正常培养的细胞作为对照组(Control);细胞H/R处理作为H/R模型组。
1.2.4 生物信息学
从GEO数据库(https://www.ncbi.nlm.nih.gov/gds/)下载GSE230165 高通量数据集。使用R软件EdgeR包分析差异表达的 miRNA,Plt;0.05设为显著差异的阈值。TargetScan7.2(http://www.targetscan.org/vert_72/)数据库预测miR-224-5p的靶基因,并对靶基因进行KEGG通路分析。
1.2.5 RNA
分离和实时荧光定量逆转录聚合酶链反应(qRT-PCR)使用TRIzol 试剂从H9c2 细胞或者200 μL血浆中提取总RNA,然后使用第一链cDNA合成试剂盒进行逆转录。对于miRNA检测,在存在茎环的情况下对1 μg总RNA进行反转录。miRNA和U6的引物由广州锐博生物技术有限公司提供。最后,根据制造商的说明,使用miRNA qRT-PCR检测试剂盒(Sparkjade)对总miRNA进行定量,U6作为内源性对照。引物序列如下:miR-224-5p, forward:5'-ACAAGTCACTAGTGGTTCC-3';reverse:5'-CAGTGATGTTGCGGTCTG-3';U6, forward: 5'-CAGCACATTCGGCAGCACA-3';reverse:5'-TGGTGTCG TTGGAGTCG-3。使用 2-△△Ct法计算miRNA的表达量。
1.2.6 噻唑蓝(MTT)检测细胞活力
用完全培养基制成细胞悬液,将细胞浓度调整至以5×104/mL,吸取100 μL稀释后的细胞悬液,加入96孔板,每组设置5个复孔,并设置对照孔。培养24 h后加入20 μL MTT(5 mg/mL)孵育4 h。然后,通过离心机去除培养基,并通过加入150 μL DMSO裂解细胞以溶解甲臜。通过酶标仪测量490 nm处的吸光度(A)来量化细胞活力,并计算细胞存活率[实验组A值/对照组A值×100%]。
1.2.7 检测氧化应激相关指标
根据试剂制造商的方案,按照试剂盒说明书,分别收集H9c2细胞培养液上清和细胞。LDH活性的检测使用LDH检测试剂盒检测各组细胞的培养液上清。将各组H9c2细胞使用细胞裂解液在冰上裂解后,收集上清液,检测SOD2 的活性和MDA的含量。
1.2.8 双荧光素酶活性测定
构建野生型(Wt)Wt-PTEN-3'-UTR和突变体(Mut)Mut-PTEN-3'-UTR荧光素酶报告基因载体,根据Lipofectamine™3000转染试剂盒说明步骤将Wt-PTEN-3'-UTR或Mut-PTEN-3'-UTR与miRNA阴性对照寡核苷酸(miRNA NC)或miR-224-5p 模拟物(miR-224-5p mimic)分别共转染至H9c2 细胞。根据转染程序,转染48 h后使用试剂盒和多模式酶标仪测量双荧光素酶报告基因活性。
1.2.9 Western blotting
收集各组H9c2细胞,用细胞裂解缓冲液裂解,并在4 ℃下以12 000 g离心15 min。用RIPA裂解缓冲液提取蛋白质,用8%~12%的分辨率凝胶进行SDS-PAGE分离。将分解后的蛋白质电印迹到膜上。在用5%脱脂牛奶封闭后。TBST溶液洗膜3次,然后分别加入稀释后的一抗:Caspase-3(1∶1000)、Bax(1∶1000)、Bcl-2(1∶1000)、PTEN(1∶1000)、Akt(1∶1000)、PI3K(1∶1000)、p-Akt(1∶2000)、p-PI3K(1∶1000)、SOD2(1∶1000)、FoxO1(1∶1000)、p-FoxO1(1∶1000)、β-actin(1∶500) 4 ℃孵育过夜。用TBST洗涤3次后,加入HRP标记的二抗IgG(1∶2000)于室温孵育1 h。使用增强化学发光显影,在Bio-Rad 化学发光成像系统下曝光,用ImageJ软件进行定量。
1.2.10 流式细胞术实验
数生长期的H9c2 心肌细胞,消化、稀释、转染、分组等操作后,用PBS 溶液进行洗涤,离心5 min后,用1×Binding Buffer缓冲液稀释成1×106/mL的细胞悬液,取100 μL细胞悬液,按照试剂盒说明书加入5 μL的Annexin V-APC和5 μL的7-AAD染色液, 轻柔涡旋混匀后,室温避光孵育20 min。,混匀后上机检测,实验重复3次。
1.3 统计学分析
SPSS 24.0、GraphPadPrism8.02 和R 语言软件进行统计分析及可视化,计量资料用均数±标准差表示。Shapiro-Wilk法用于检验正态性,Levene法用于评估方差齐性。各组间差异比较采用单因素方差分析两两比较用LSD法,两组比较采用独立样本t检验;计数资料采用Fisher精确检验或χ2检验。此外,受试者工作特征曲线(ROC)下面积(AUC)被用来评估miRNA 作为AMI 的诊断标志物的准确性。Plt;0.05(双侧)时,差异被认为具有统计学意义。
2 结果
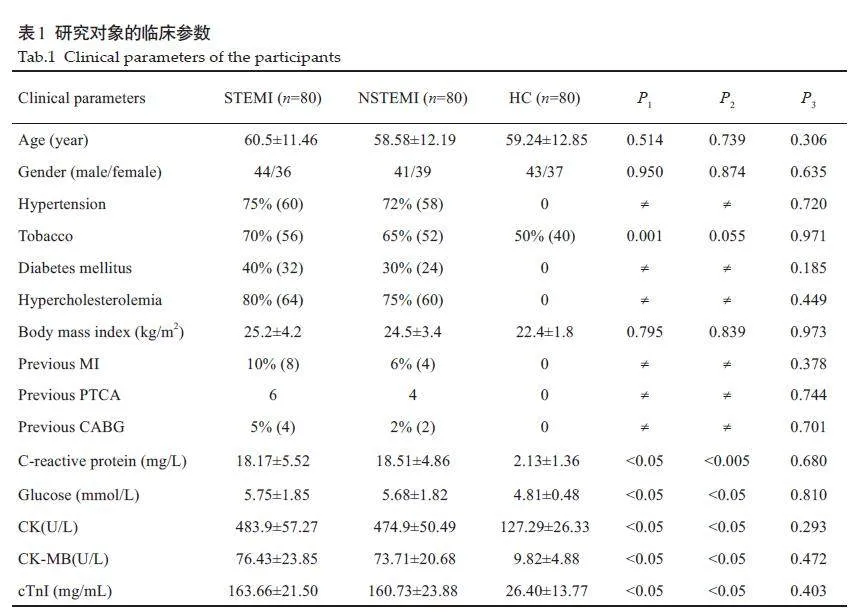
2.1 研究对象的临床参数
80例STEMI患者和80例NSTEMI患者,与健康对照组(HC)在年龄和性别完全匹配,基本信息和实验室检测结果如表1所示。STEMI和NSTEMI患者的血糖、C反应蛋白、CK、CK-MB和cTnI水平均显著高于对照组(Plt;0.05)。
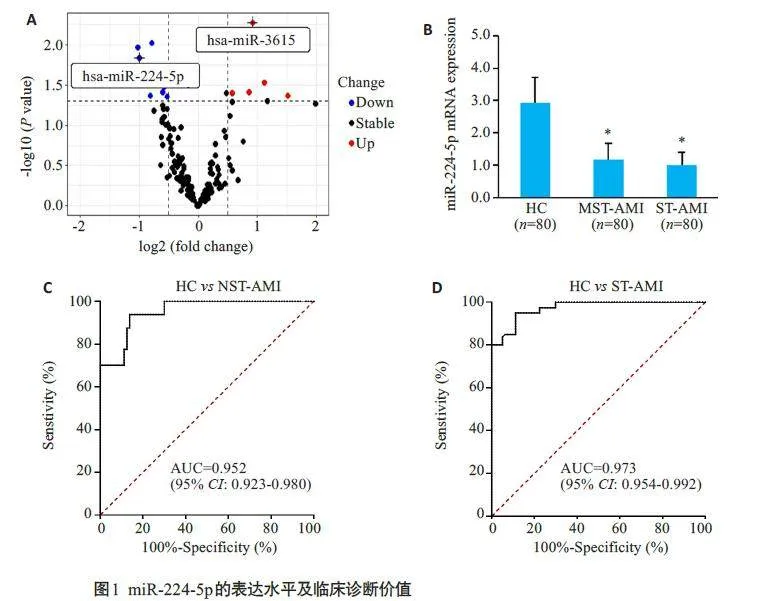
2.2 miR-224-5p在AMI疾病中的表达水平
高通量数据分析显示miR-224-5p显著下调(图 1A)。AMI患者与HC组血浆miR-224-5p表达水平结果显示STEMI(n=80)和NSTEMI(n=80)组血浆miR-224-5p的水平均低于HC组,差异有统计学意义(Plt;0.05)。但是STEMI 与NSTEMI 患者血浆中miR-224-5p 的水平差异无统计学意义(Pgt;0.05,图1B);miRNA-224-5p 在AMI 患者和健康受试者之间表现出很强的分离能力,STEMI的曲线下面积(AUC)为0.952(图1C);NSTEMI患者和健康被试者的AUC为0.973(Plt;0.001,图1D)。
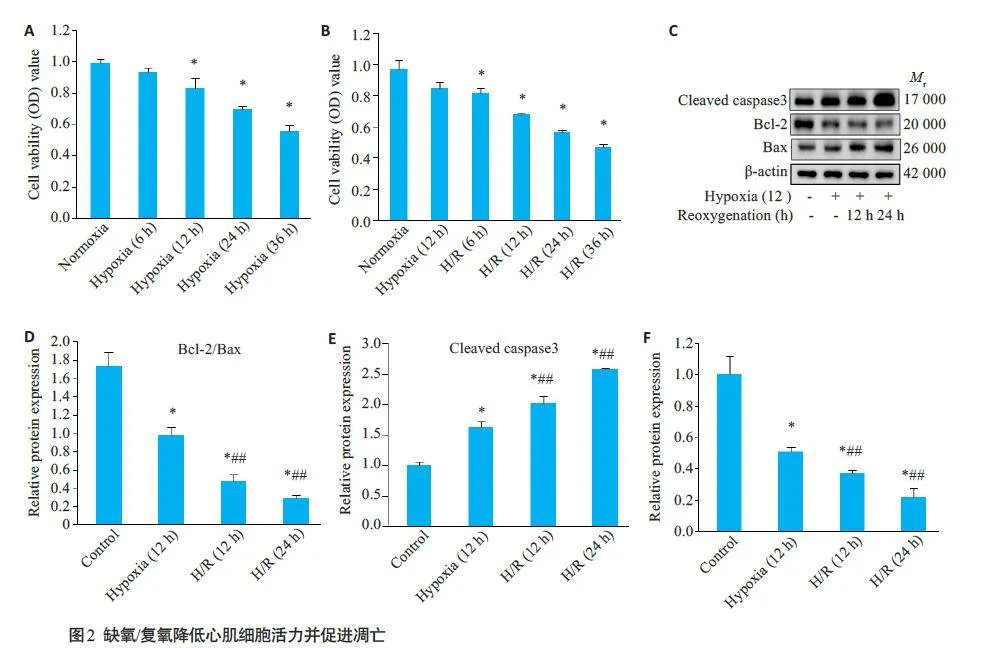
2.3 缺氧/复氧降低心肌细胞活力
缺氧引起心肌细胞活力的时间依赖性下降,缺氧暴露后12 h开始显著下降(Plt;0.05,图2A);随着复氧时间的增加,在12 h复氧后,心肌细胞活力也显著降低(Plt;0.05,2B);Western blotting结果显示,与对照组相比,低氧暴露12 h 后,心肌细胞凋亡活性显著增加,表现为Bcl-2/Bax比值降低,Cleaved caspase3表达升高;随后复氧时间的延长心肌细胞Bcl-2/Bax 蛋白比值逐渐降低、Cleaved caspase3 的表达上升,缺氧/复氧降低了细胞存活率(Plt;0.05,图2C~E);在H/R 处理的H9c2 细胞中,miR-224-5p的表达水平明显降低(Plt;0.05,图2F)。
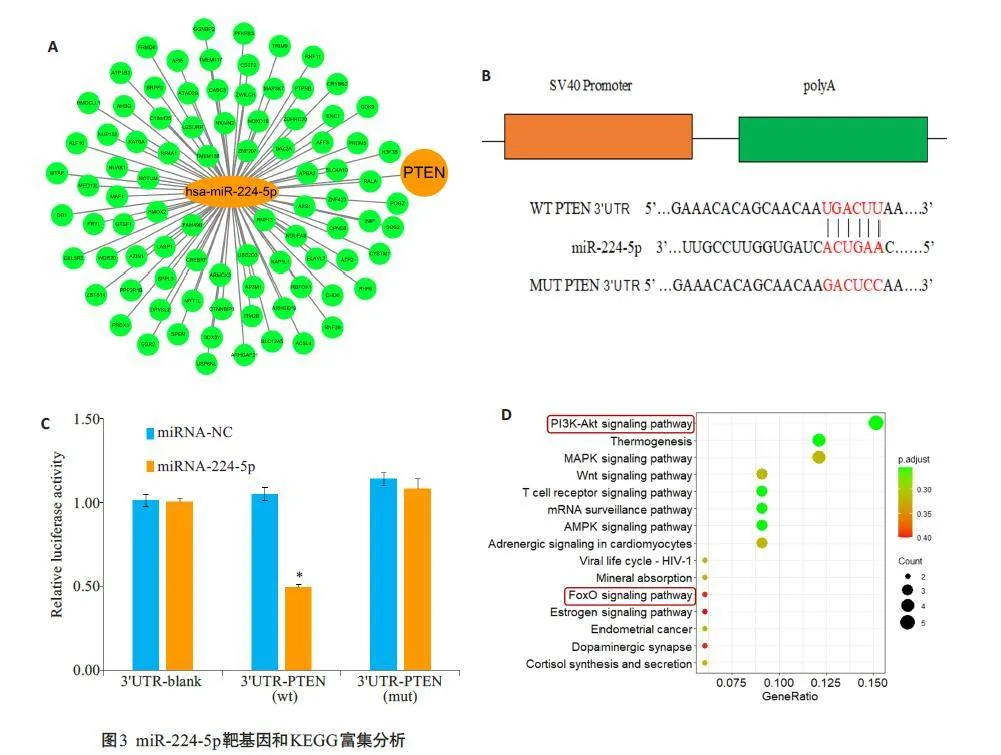
2.4 miR-224-5p靶向调控PTEN的表达
基于在线数据库筛选miR-224-5p的靶基因(图3A)。TargetScan数据库的分析显示,miR-224-5p与PTEN之间存在结合位点(图3B)。双荧光素酶报告基因实验显示,在H/R处理的H9c2 细胞中,我们发现miR-224-5pmimic显著抑制野生型PTEN 3'-UTR报告基因的荧光素酶活性,差异有统计学意义(Plt;0.05),但不影响具有突变miR-224-5p结合位点的PTEN 3'-UTR报告基因的荧光素酶活性(Pgt;0.05)。此外,当miR-NC或Vector与Wt或Mut共转染时,组间荧光素酶活性没有变化无统计学意义(Pgt;0.05,图3C)。京都基因和基因组百科全书(KEGG)对预测的靶基因进行的信号通路分析表明PI3K/Akt是最显著富集的通路(图3D)。
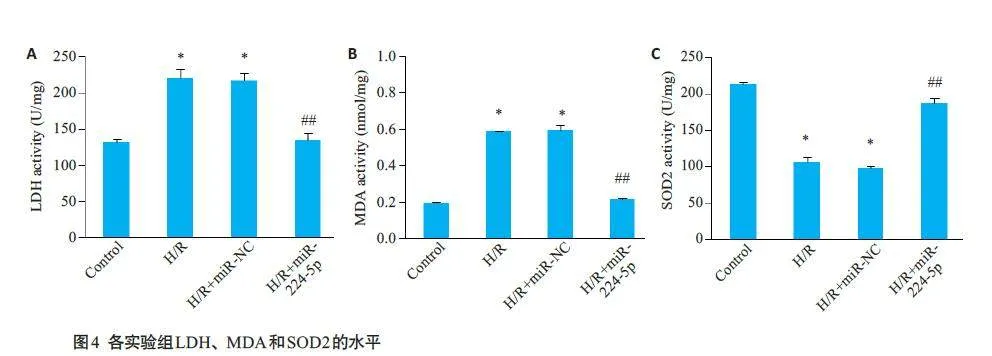
2.5 各实验组SOD2、LDH及MDA水平比较
与Control 组比较,H/R组上清中SOD2 的水平降低,MDA和LDH水平增加(Plt;0.05);与H/R+miR-NC组比较,H/R+miR-224-5P组SOD2的水平上调,MDA和LDH的水平显著下降(Plt;0.05);H/R+miR-NC组与H/R组各指标比较差异均无统计学意义(Pgt;0.05,图4)。
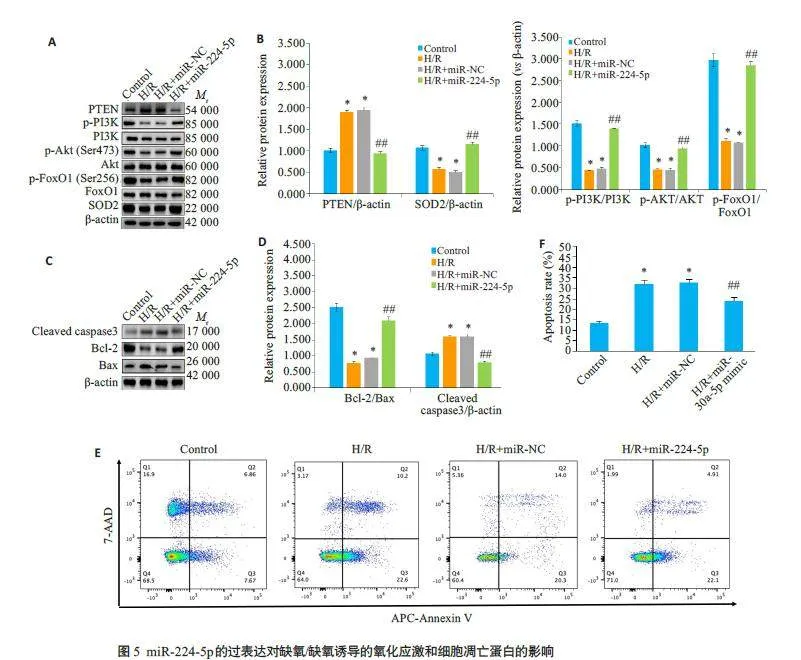
2.6 miR-224-5p 通过PI3K/Akt/FoxO1 信号传导参与H/R诱导的心肌细胞损伤
与Control 组相比,H/R 组细胞中PTEN、Cleavedcaspase-3 和bax 蛋白表达水平明显上调(Plt;0.05),p-PI3K、p-Akt、p-FoxO1、SOD2 和Bcl-2 蛋白表达水平以及Bcl-2/Bax比值显著下调(Plt;0.05);与H/R+miR-NC组比较,H/R+miR-224-5p mimic 组细胞中PTEN、Cleaved caspase-3 和Bax 蛋白表达水平均显著降低(Plt;0.05),p-PI3K、p-Akt、p-FoxO1、SOD2 和Bcl-2 蛋白表达水平及Bcl-2/Bax比值显著升高(Plt;0.05,图5)。流式细胞术结果显示,与Control 组相比,H/R 组的细胞凋亡率显著升高(Plt;0.05);与H/R+miR-NC组相比,H/R+miR-224-5p mimic组的细胞凋亡率降低(Plt;0.05,图5E、F)。
3 讨论
缺血/再灌注损伤是缺血性心脏病的主要病因,因此减轻心肌细胞损伤并因此保护心脏功能对于降低心血管疾病风险至关重要[20]。研究表明miRNAs 在心肌梗死中异常表达并在调节心肌损伤中发挥作用,但miRNAs 在心肌梗死中的功能仍未完全了解。本研究探究了miR-224-5p在AMI患者中的诊断价值,与健康对照组相比,STEMI和NSTEM患者miRNA-224-5p血浆中浓度显著降低,miRNA-224-5p的ROC曲线下面积(AUC)在HC 与STEMI(AUC=0.973)和NSTEMI(AUC=0.952)表现出很强的区分性,进一步在细胞上研究对缺氧/复氧的影响。MTT的结果显示缺氧/复氧处理降低了细胞活力,激活了细胞中的凋亡途径。细胞凋亡是缺血/再灌注损伤的严重后果,当凋亡途径被激活时,会发生各种变化,包括细胞收缩和脱落、核碎裂和凋亡体的形成[21]。在凋亡相关蛋白中,Bcl-2家族和裂解的caspase-3是调控细胞凋亡的关键因子,Bcl-2可能促进细胞存活,而Bax可能通过凋亡促进细胞死亡。Bcl-2/Bax 比值是决定细胞损伤后是否发生细胞凋亡的关键因素[22]。Western blotting结果显示,与对照组相比,H/R可显著下调Bcl-2/Bax 的比值,上调Cleaved caspase-3蛋白的表达。此外越来越多的研究表明在缺氧/复氧诱导的心肌细胞损伤过程中miR-7a-5p、miR-1224 和MicroRNA-590-3p的浓度发生了严重的变化。经特异性抑制或模拟miRNAs处理的H/R诱导的H9c2细胞通过作用其靶基因(分别为VDAC1、HIF-1α和GPX4)显著调节各种细胞炎症途径,显著降低氧化应激、caspase-3活性和细胞凋亡,并显著保护心肌细胞损伤提高细胞存活率[23-25]。在此,我们发现miR-224-5p 在缺氧/再灌注后H9c2细胞中也显著下调。然而,miR-224-5p对H/R暴露的心肌细胞损伤中氧化应激和凋亡的报道较少。
本研究探讨了H/R诱导的H9c2 细胞miR-224-5p水平显著降低的潜在分子机制。使用TargetScan在线数据库预测miR-224-5p 可能的靶基因,发现miR-224-5p 与PTEN的3'非翻译区(3'-UTR)存在特异的结合位点。荧光素酶报告基因实验证实PTEN是miR-224-5p的靶基因。PTEN作为一种脂质磷酸酶,负调控PI3K信号通路并将PIP3转化为PIP2。当PTEN发生突变或失活时,PI3K效应子特别是Akt,在没有任何外源致癌刺激的情况下被激活。PI3K/Akt信号通路参与多种生理过程,是许多疾病尤其是肿瘤发展的重要信号途径,能调控细胞存活、转移和新陈代谢,在凋亡和炎症因子募集中发挥作用[26, 27]。如Akt 磷酸化Bcl-2 家族成员BAD,使其与14-3-3结合而阻止其与Bcl-XL结合起始凋亡[28]。PTEN/PI3K/Akt-mTOR信号通路是缺血性心脏病中BMI1 诱导的心脏纤维化的原因[29]。KEGG富集分析显示miR-224-5p的预测靶基因与最显著富集的通路PI3K/Akt通路相关。研究表明,PI3K/Akt通路在抵抗ROS 方面发挥关键作用,并改善心肌细胞的存活[30]。叉头盒转录因子O1(FoxO1)是一种调节抗氧化反应的重要转录因子,已被认为是改善心脏氧化应激损伤的有趣靶点[31]。FoxO1又是PI3K/Akt信号通路的下游靶标。氧化应激是ROS生成和抗氧化防御系统之间失衡的结果,乳酸脱氢酶和丙二醛是细胞氧化应激的重要指标,而超氧化物歧化酶是保护性指标[32]。本研究发现,与对照组相比,H9c2 细胞进行H/R 处理后,细胞LDH和MDA水平升高,同时SOD2 表达也减少,表明H/R后确实能够使细胞发生氧化应激,抗氧化系统受损。与H/R组比较,miR-224-5p mimic转染H/R诱导的H9c2细胞后,LDH的活性降低,MDA含量减少而SOD2活力上升,结果表明,miR-224-5p可显著抑制H/R暴露的心肌细胞的氧化应激,提示miR-224-5p 可能通过阻断氧化应激而对H/R处理的心肌细胞产生抗凋亡作用。
为了研究miR-224-5p 是否通过PI3K/Akt/FoxO1抑制氧化应激减少细胞凋亡。体外模拟miR-224-5p过表达,WB结果显示,与Control组相比,H/R组H9c2细胞中PTEN、Cleaved caspase-3 和Bax 蛋白表达水平升高,p-Akt、p-FoxO1、SOD2 和Bcl-2 蛋白表达水平以及Bcl-2/Bax 比值降低。miR-224-5p 过表达后p-PI3K、p-Akt、p-FoxO1、SOD2和Bcl-2蛋白的表达水平以及Bcl-2/Bax比值均明显升高,PTEN、Cleaved caspase-3和bax蛋白表达水平显著降低。此外,流式细胞术结果显示miR-224-5p 转染减少了H/R引起的H9c2 心肌细胞凋亡。表明miR-224-5p过表达可显著减轻氧化应激和抑制细胞凋亡,从而保护细胞免受H/R的影响。
综上,本研究发现miR-224-5p 保护心肌细胞免受缺氧/复氧损伤与PI3K/Akt/FoxO1信号通路密切相关。然而,本研究仅在体外细胞水平上研究了miR-224-5p在H/R中的机制。为了更准确地分析miR-224-5p调控PI3K/Akt/FoxO1信号通路在AMI中的机制,并为未来的临床应用提供思路,需要在体内实验上进行进一步的验证性研究。
参考文献:
[1] WHO CVD Risk Chart Working Group. World Health Organizationcardiovascular disease risk charts: revised models to estimate risk in21 global regions[J]. Lancet Glob Health, 2019, 7(10): e1332-e1345.
[2] Valikeserlis I, Athanasiou AA, Stakos D. Cellular mechanisms andpathways in myocardial reperfusion injury[J]. Coron Artery Dis,2021, 32(6): 567-77.
[3] Liu Y, Li L, Wang Z, et al. Myocardial ischemia-reperfusion injury;Molecular mechanisms and prevention[J]. Microvasc Res, 2023,149: 104565.
[4] Zhang WL, Chen CY, Wang J, et al. Mitophagy in cardiomyocytesand in platelets: a major mechanism of cardioprotection againstischemia/reperfusion injury[J]. Physiology, 2018, 33(2): 86-98.
[5] Wang L, Niu HP, Zhang J. Homocysteine induces mitochondrialdysfunction and oxidative stress in myocardial ischemia/reperfusioninjury through stimulating ROS production and the ERK1/2signaling pathway[J]. Exp Ther Med, 2020, 20(2): 938-44.
[6] Jiang W, Zhang YX, Zhang W, et al. Hirsutine amelioratesmyocardial ischemia-reperfusion injury through improvingmitochondrial function via CaMKII pathway[J]. Clin ExpHypertens, 2023, 45(1): 2192444.
[7] Zhao P, Zhang BL, Liu K, et al. Overexpression of miR-638attenuated the effects of hypoxia/reoxygenation treatment on cellviability, cell apoptosis and autophagy by targeting ATG5 in thehuman cardiomyocytes[J]. Eur Rev Med Pharmacol Sci, 2018, 22(23): 8462-71.
[8] Zhai CL, Sun YG, Qian G, et al. LncRNA AK087124/miR-224-5p/PTEN axis modulates endothelial cell injury in atherosclerosisthrough apoptosis and AKT signaling pathway[J]. Arch BiochemBiophys, 2021, 705: 108916.
[9] Mao CY, Zhang TT, Li DJ, et al. Extracellular vesicles from hypoxiapreconditionedmesenchymal stem cells alleviates myocardial injuryby targeting thioredoxin-interacting protein-mediated hypoxiainduciblefactor-1α pathway[J]. World J Stem Cells, 2022, 14(2):183-99.
[10]Cheng JW, Ma HB, Yan M, et al. Circ_0007624 suppresses the development of esophageal squamous cell carcinoma via targetingmiR-224-5p/CPEB3 to inactivate the EGFR/PI3K/AKT signaling[J]. Cell Signal, 2022, 99: 110448.
[11] Zhou G, Wu H, Yang J, et al. Liraglutide attenuates myocardialischemia/reperfusion injury through the inhibition of necroptosis byactivating GLP-1R/PI3K/akt pathway[J]. Cardiovasc Toxicol,2023, 23(3/4): 161-75.
[12]Guo LT, Wang SQ, Su J, et al. Baicalin amelioratesneuroinflammation-induced depressive-like behavior throughinhibition of toll-like receptor 4 expression via the PI3K/AKT/FoxO1 pathway[J]. J Neuroinflammation, 2019, 16(1): 95.
[13]Zeng H, Yang Y, Tou FF, et al. Bone marrow stromal cell-derivedexosomes improve oxidative stress and pyroptosis in doxorubicininducedmyocardial injury in vitro by regulating the transcription ofGSDMD through the PI3K-AKT-Foxo1 pathway[J]. ImmunInflamm Dis, 2023, 11(3): e810.
[14]Tan Y, Bie YL, Chen L, et al. Lingbao Huxin pill alleviates apoptosisand inflammation at infarct border zone through SIRT1-mediatedFOXO1 and NF- κB pathways in rat model of acute myocardialinfarction[J]. Chin J Integr Med, 2022, 28(4): 330-8.
[15]Liu LL, Qiao S, Wang ML, et al. MiR224-5p inhibitor restrainsneuronal apoptosis by targeting NR4A1 in the oxygen-glucosedeprivation (OGD) model[J]. Front Neurosci, 2020, 14: 613.
[16]Hou ZX, Qin XH, Hu YY, et al. Longterm exercise-derivedexosomal miR-342-5p: a novel exerkine for cardioprotection[J].Circ Res, 2019, 124(9): 1386-400.
[17]Gao Y, Yin H, Zhang YF, et al. Dexmedetomidine protectshippocampal neurons against hypoxia/reoxygenation-inducedapoptosis through activation HIF-1α/p53 signaling[J]. Life Sci,2019, 232: 116611.
[18]Thygesen K, Alpert JS, Jaffe AS, et al. Fourth universal definition ofmyocardial infarction (2018)[J]. Eur Heart J, 2019, 40(3): 237-69.
[19]Lv XB, Niu QH, Zhang M, et al. Critical functions of microRNA-30a-5p-E2F3 in cardiomyocyte apoptosis induced by hypoxia/reoxygenation[J]. Kaohsiung J Med Sci, 2021, 37(2): 92-100.
[20]Wu MY, Yiang GT, Liao WT, et al. Current mechanistic concepts inischemia and reperfusion injury[J]. Cell Physiol Biochem, 2018, 46(4): 1650-67.
[21]Monier B, Suzanne M. Orchestration of force generation and nuclearcollapse in apoptotic cells[J]. Int J Mol Sci, 2021, 22(19): 10257.
[22]Zhu L, Hao J, Cheng MJ, et al. Hyperglycemia-induced Bcl-2/Bax-mediated apoptosis of Schwann cells via mTORC1/S6K1 inhibitionin diabetic peripheral neuropathy[J]. Exp Cell Res, 2018, 367(2):186-95.
[23]Lu HL, Zhang JF, Xuan FF. MiR-7a-5p attenuates hypoxia/reoxyenation-induced cardiomyocyte apoptosis by targetingVDAC1[J]. Cardiovasc Toxicol, 2022, 22(2): 108-17.
[24]Li GB, Jin JL, Liu SX, et al. Inhibition of miR-1224 suppresseshypoxia/reoxygenation-induced oxidative stress and apoptosis incardiomyocytes through targeting GPX4[J]. Exp Mol Pathol, 2021,121: 104645.
[25]Gong NJ, Yang XH, Li XY, et al. MicroRNA-590-3p relieveshypoxia/reoxygenation induced cardiomyocytes apoptosis andautophagy by targeting HIF-1α[J]. Exp Ther Med, 2021, 22(4):1077.
[26]高晓阳, 赵晓璐, 张春艳, 等. 槲皮素诱导肝星状细胞凋亡: 基于调控miR-146影响PI3K/Akt信号通路[J]. 南方医科大学学报, 2023, 43(10): 1725-33.
[27]杨 子, 赵天豪, 程 阳, 等. 香叶木素通过调节小鼠的肠道免疫平衡减轻克罗恩病样结肠炎:基于抑制PI3K/AKT通路[J]. 南方医科大学学报, 2023, 43(3): 474-82.
[28]Xu YX, Jiang YP, Wang YZ, et al. LINC00473 rescues human bonemarrow mesenchymal stem cells from apoptosis induced bydexamethasone through the PEBP1-mediated Akt/Bad/Bcl-2signaling pathway[J]. Int J Mol Med, 2021, 47(1): 171-82.
[29]Yang WB, Wu ZJ, Yang K, et al. BMI1 promotes cardiac fibrosis inischemia-induced heart failure via the PTEN-PI3K/Akt-mTORsignaling pathway[J]. Am J Physiol Heart Circ Physiol, 2019, 316(1): H61-H69.
[30]Sun L, Wang H, Xu D, et al. Lapatinib induces mitochondrialdysfunction to enhance oxidative stress and ferroptosis indoxorubicin-induced cardiomyocytes via inhibition of PI3K/AKTsignaling pathway[J]. Bioengineered, 2022, 13(1): 48-60.
[31]Peng B, Rao L, Yang JL, et al. Columbianadin attenuatesdoxorubicin-induced cardiac injury, oxidative stress, and apoptosisvia Sirt1/FOXO1 signaling pathway[J]. Acta Cir Bras, 2023, 38:e382223.
[32]Hu B, Tian T, Li XT, et al. Dexmedetomidine postconditioningattenuates myocardial ischemia/reperfusion injury by activating theNrf2/Sirt3/SOD2 signaling pathway in the rats[J]. Redox Rep,2023, 28(1): 2158526.
(编辑:吴锦雅)
基金项目:河北省政府资助临床医学人才培养项目(2020009);河北省自然科学基金资助项目(H2024307005)
猜你喜欢
世界科学技术-中医药现代化(2021年7期)2021-11-04 08:10:24
中成药(2018年5期)2018-06-06 03:11:46
右江医学(2016年5期)2017-05-06 12:12:03
新教育时代·教师版(2017年2期)2017-03-23 12:04:55
中国医药导报(2017年3期)2017-03-21 21:54:07
中国医药导报(2016年32期)2017-02-28 17:00:36
中国现代医生(2016年23期)2016-11-15 02:47:50
中国实用医药(2016年17期)2016-07-26 14:15:36
中国药理学与毒理学杂志(2015年3期)2015-12-16 09:11:40
医学信息(2015年15期)2015-07-07 14:59:00
