
左氧氟沙星薄膜衣片的制备及其质量评价
2024-11-01 00:00张震陈亚军龚江雪张兰
武汉纺织大学学报 2024年5期


摘要:自研制剂和原研制剂的质量一致可以较大程度地保证二者临床效果一致。因此,以颗粒成型、脆碎度、体外溶出度等为评价指标,通过试验对润湿剂种类、填充剂和崩解剂、润滑剂、粘合剂和包衣粉等辅料的用量进行筛选及优化自研制剂的处方。经试验选择最优处方:左氧氟沙星512.5mg、微晶纤维素70mg、羧甲纤维素钙30mg、羟丙纤维素3.75mg、硬脂富马酸钠27mg。包衣增重选择3~5%范围。对优选处方制备的薄膜衣片进行质量检查,并与原研制剂进行对比,在磷酸盐缓冲(pH6.8)溶液、盐酸溶液(pH1.0)以及水和醋酸盐缓冲(pH4.5)溶液这四种不同的溶出介质中,自研产品和原研产品都可以15min达到85%以上的溶出度,具有相似的溶出行为;且它们的晶体结构和含量都是一致的。此外,经过6个月的稳定性加速试验,这些产品的检测结果没有发生任何明显的改变。证明自研左氧氟沙星片的处方及工艺稳定可行,与原研制剂具有质量一致性。
关键词:左氧氟沙星;溶出度;稳定性;质量一致性
中图分类号:R944.4文献标志码:A文章编号:2095-414X(2024)05-0071-08
0引言
左氧氟沙星(levofloxacin)属于第三代喹诺酮类的临床常用的广谱抗菌药,属于浓度依赖型[1],通过抑制细菌的拓扑异构酶和脱氧核糖核酸(DNA)回旋酶活性,使得细菌DNA不能增殖[2],从而发挥很好的杀菌作用,适用于各种敏感细菌引起的感染疾病的治疗[3]。左氧氟沙星薄膜衣片(商品名:可乐必妥,规格:0.5g)由日本第一三共药品有限公司研究开发,于2004年在国内首先上市。本研究选择第一三共制药(北京)有限公司上市的原研地产化产品左氧氟沙星片作为参比制剂进行仿制,并使用体外溶出度方法来评价自制制剂与原研制剂的体外溶出相似性[4],结合含量和加速稳定性试验评价自制产品与原研产品的质量一致性[5],是对于自制产品的处方和生产技术工艺开发的支持。
1实验部分
1.1实验仪器
RC8MD及RZQ-8D溶出度测试仪,由天大天发公司提供;TU-1810紫外可见分光光度计,由岛津企业管理(中国)有限责任公司提供;waters2695-2487液相仪,由沃特世科技(上海)有限责任公司提供;JHY-500A快速水分仪,由福建米德电子科技公司提供;PG20高速旋转式压片机,由北京新龙立公司提供;SQP SECURA225D-1CN电子天平,德国塞多利丝科学仪器公司产品;CHC-48型高效包衣机,创志机电科技公司的设备。
1.2实验材料
左氧氟沙星片原研制剂(批号:BW052G1,BW056G1,BW069G1,第一三共药品公司生产);左氧氟沙星对照品(中检所供应,批号:130455-201607,含量97.3%);氧氟沙星对照品(批号:130454-201206,中检所供应,含量99.5%);左氧氟沙星原料药(批号:KY-LFA-20181105H,KY-LFA-20171201H,浙江普洛康裕制药有限公司,含量:100.8%);微晶纤维素(批号:C1802062,C1909026,明台化工工厂生产);羧甲纤维素钙[斯百全化学(上海)有限公司,批号:3HC0034,201908095];羟丙纤维素(Ashland Specialty Ingredients G.P.,批号:177012,188444,192919);乙醇(湖南湘易康制药有限公司,批号:100320191110,100320200310);硬脂富马酸钠(MOEHS CANTABRA S.L.,批号:1863,1972,2060,2062,2368);药用薄膜包衣预混辅料(胃溶型)(批号:WXL1911055,WXL2007024,温州小伦包衣技术有限公司)。
1.3左氧氟沙星薄膜衣片的制备
称取处方量经过60目筛网的左氧氟沙星原料药,将其与微晶纤维素、羧甲纤维素钙进行混合后,并加入羟丙纤维素乙醇溶液,然后使用快速搅拌湿法制粒机制备出软材,接着将其经过20目药典筛网制湿颗粒,并在流化床干燥机中50℃烘干,最终将其经过整粒,再加入称取好的润滑剂硬脂富马酸钠,混合均匀后,用17*8.5mm椭圆冲压片,再以胃溶型药用薄膜包衣预混辅料进行薄膜包衣,制备左氧氟沙星薄膜衣片。
1.4溶出度测定方法
根据《日本药典》第17版中左氧氟沙星片溶出度测试方法和《人体生物等效性试验豁免指导原则》的要求,确定溶出考察方法[6]为:第二法(桨法),转速:50r·min-1,水浴温度:(37±0.5)℃,介质:磷酸盐缓冲(pH6.8)溶液、盐酸(pH1.0)溶液、醋酸盐缓冲(pH4.5)溶液和水,溶出介质的体积:500ml。
1.5左氧氟沙星薄膜衣片处方筛选与优化
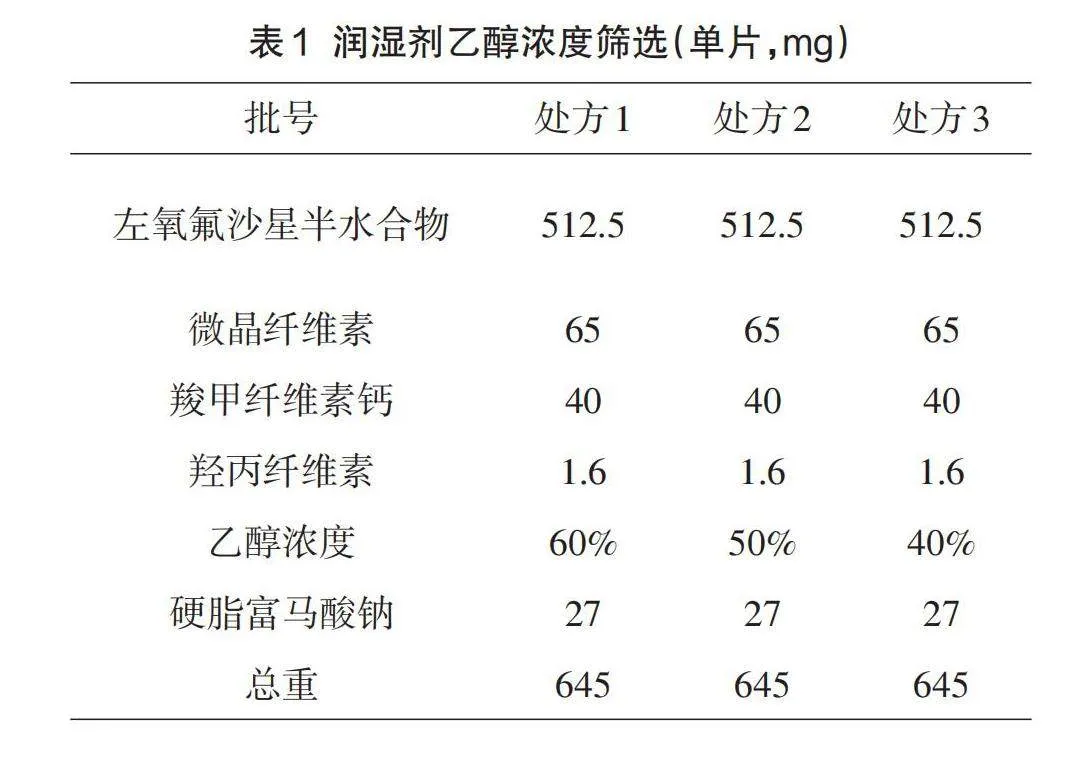
1.5.1润湿剂乙醇浓度的筛选
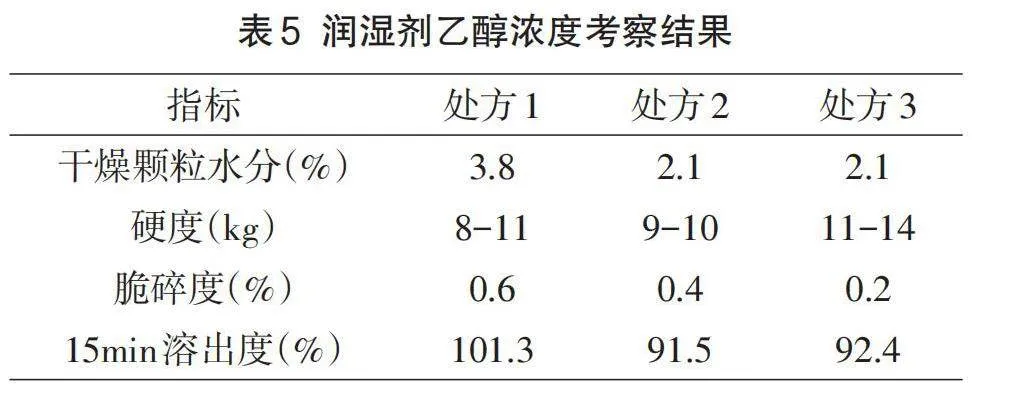
初步研究中发现左氧氟沙星原料药为半水合物,在水中溶解度较好,容易被润湿剂诱发自身粘性[7],导致制粒过程可控性差,所以选择乙醇溶液做润湿剂,对乙醇浓度进行筛选。设计处方1-3,按1.3项下制备工艺进行制备,具体见表1。对各处方进行干燥颗粒水分、素片硬度、脆碎度、溶出度试验考察。
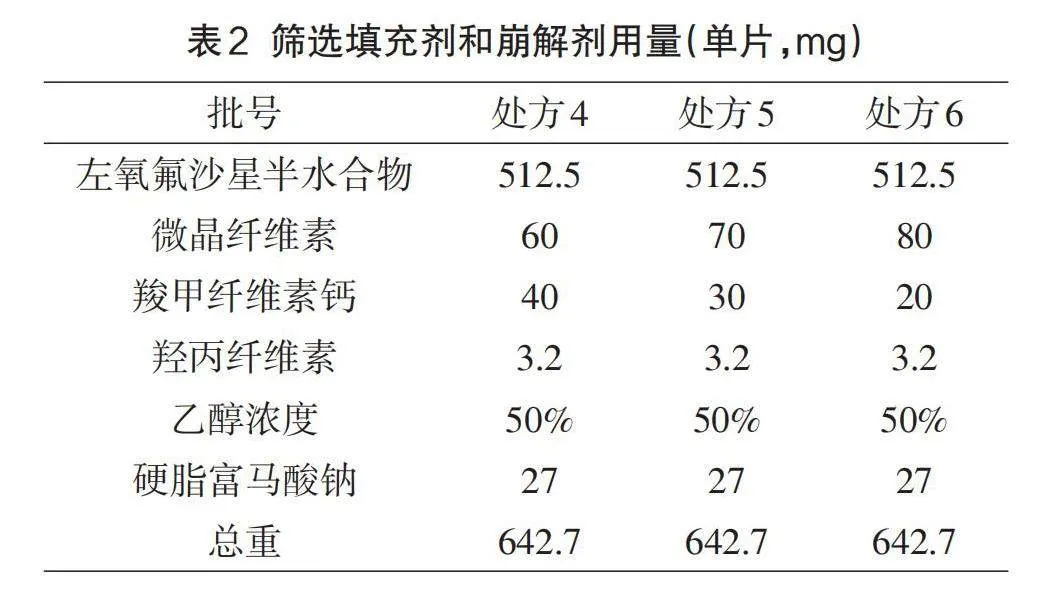
1.5.2崩解剂和填充剂的筛选
本品所用的填充剂为白色无臭无味的微晶纤维素[8],是具有多孔结构的晶型粉末,它具有出色的可压缩性和成型性,并且能够很好地与其他物料结合,使用它压制的片剂硬度也很高。原研所用崩解剂为羧甲纤维素[9]因国内暂无该产品上市,选择同类产品羧甲纤维素钙为崩解剂,二者性质接近,稳定性良好。经过对原研片剂产品细致的分析,筛选本品选用的微晶纤维素和羧甲纤维素钙的最佳用量,制定处方4-6,按1.3项下制备工艺进行制备,详情请参见表2。以制备软材情况、成颗粒情况、素片表面、崩解时限和溶出度作为考察指标。
1.5.3粘合剂的筛选
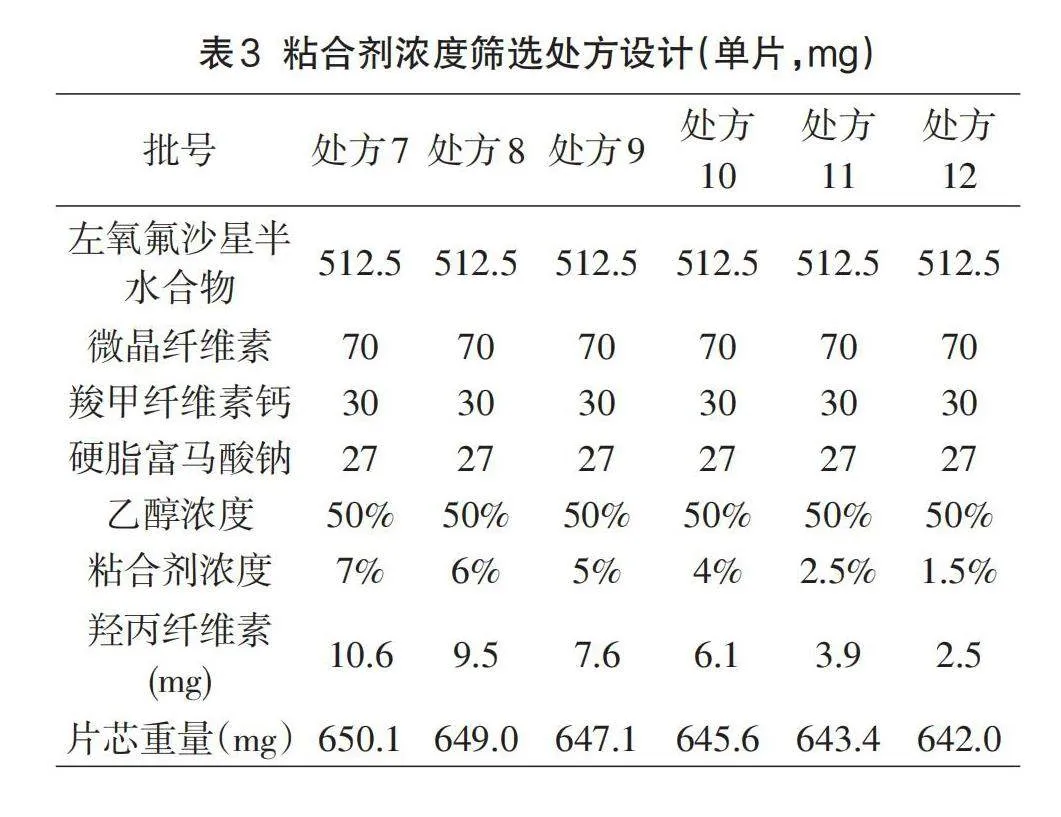
试验表明,羟丙纤维素对片剂的硬度、脆碎度、崩解、体外溶出有影响。当羟丙纤维素的添加量增多时,其硬度有增加的趋势,脆碎度有降低的趋势,但它的体外溶出和崩解过程会变得更慢。这很可能是由于羟丙纤维素是一种黏结剂,添加量越多,颗粒之间的黏结力就越强,导致压制的片剂硬度上升,脆碎度也会下降,但会导致片芯的崩解过程变得更慢,同时也致使药物从片剂中的溶出速度变慢,制定处方7-12,按1.3项下制备工艺进行制备,详情参见表3。对制定的各处方压制药片的硬度、脆碎度、溶出度进行考察。
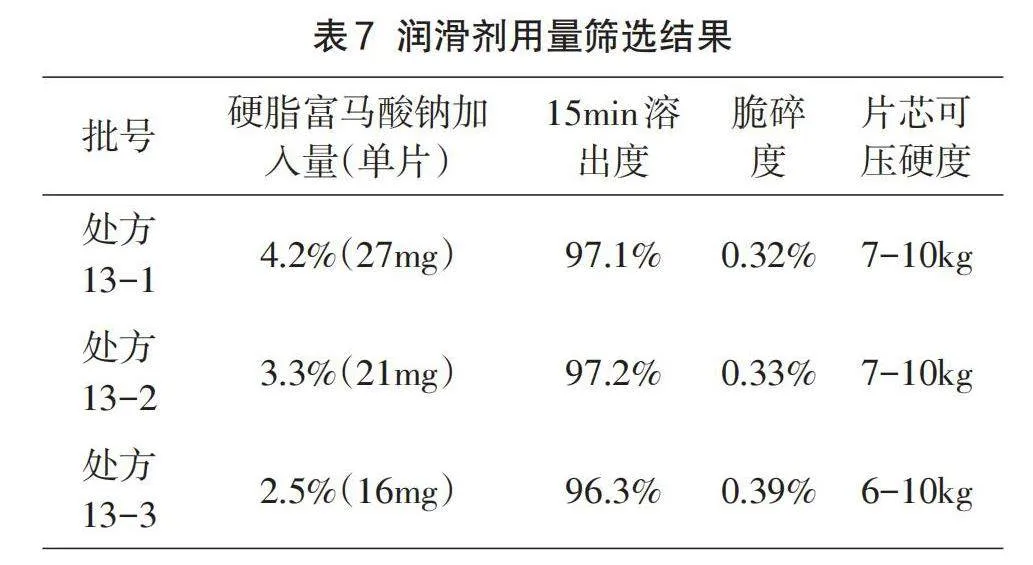
1.5.4润滑剂用量筛选
本品采用硬脂富马酸钠作为润滑剂,实验采用同一批颗粒,物料中分别加入4.2%(27mg)、3.3%(21mg)、2.5%(16mg)进行总混,详情请参考表7。对不同硬脂富马酸钠加入量压制药片的硬度、脆碎度、溶出度进行考察。
1.5.5包衣厚度筛选
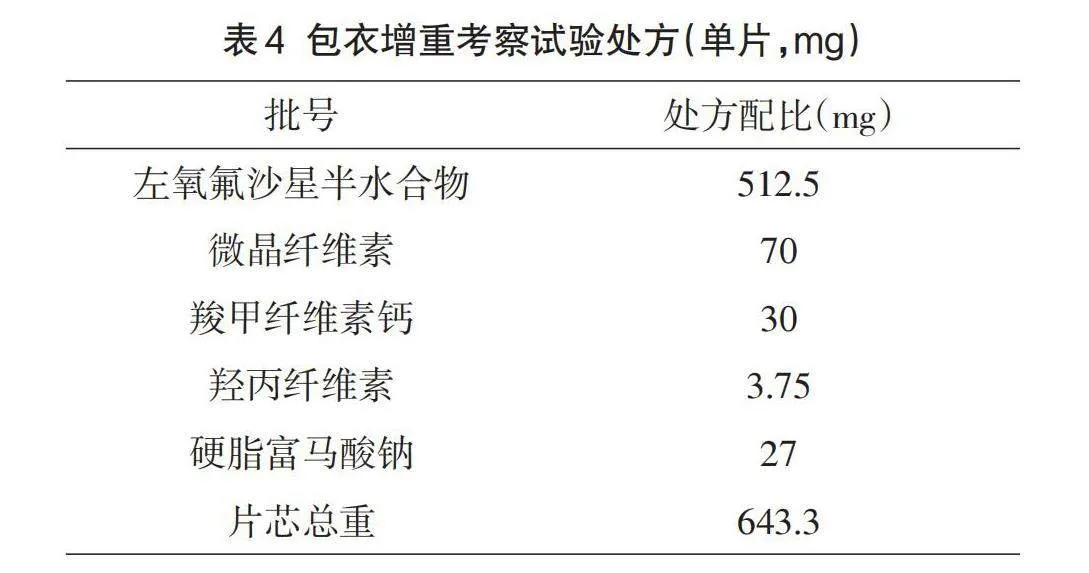
根据上述左氧氟沙星薄膜衣片的处方筛选结果,确定了本产品处方构成,具体组成请参见表4。本品为速释片,根据常规包衣经验,制备1000片左氧氟沙星素片,并使用胃溶型药用薄膜包衣预混辅料将其薄膜包衣,分别取增重约3%、4%和5%的包衣片,考察包衣前后溶出变化以及体外溶出与不同包衣厚度的关系。
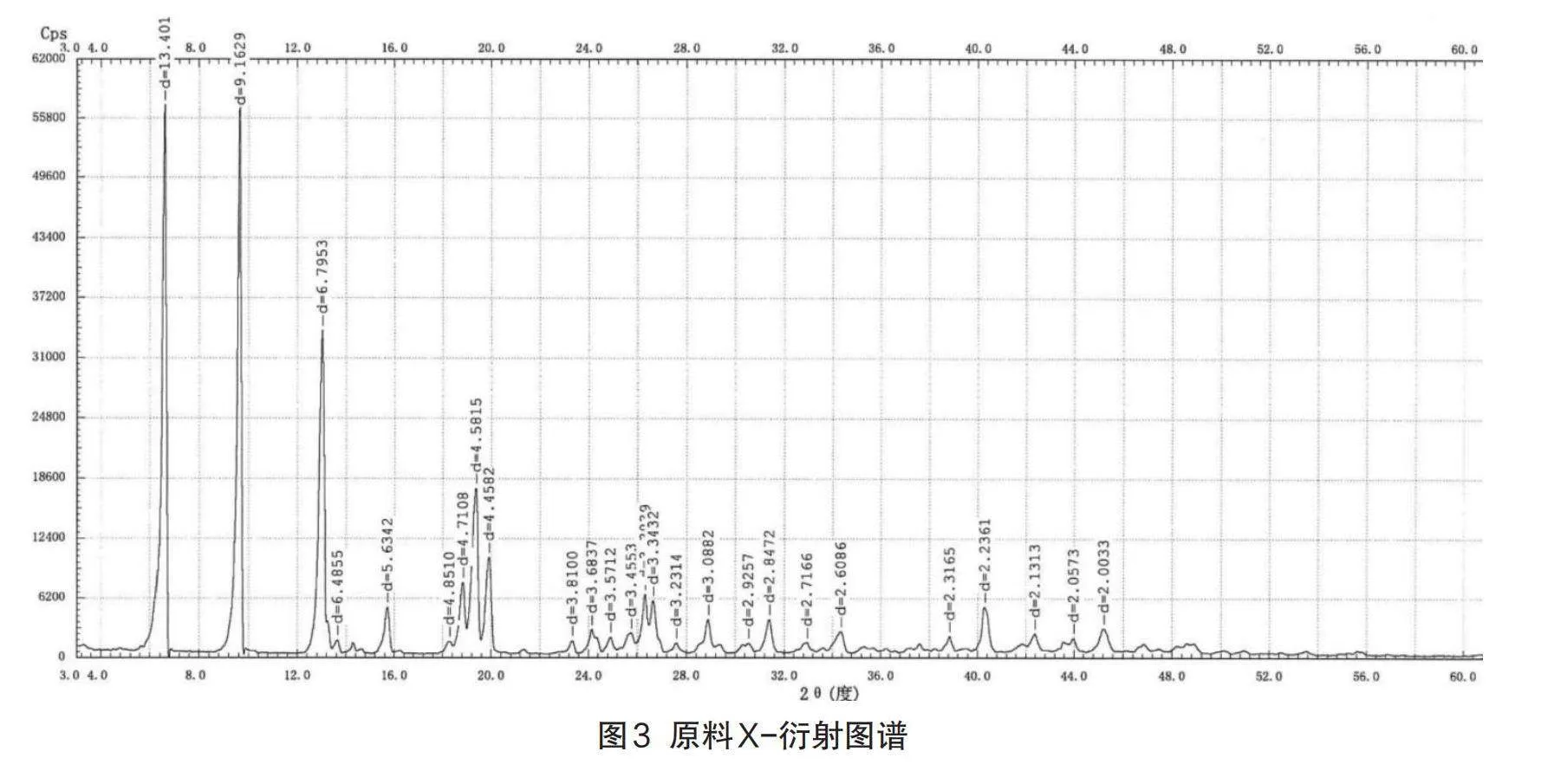
1.5.6原料药晶型研究
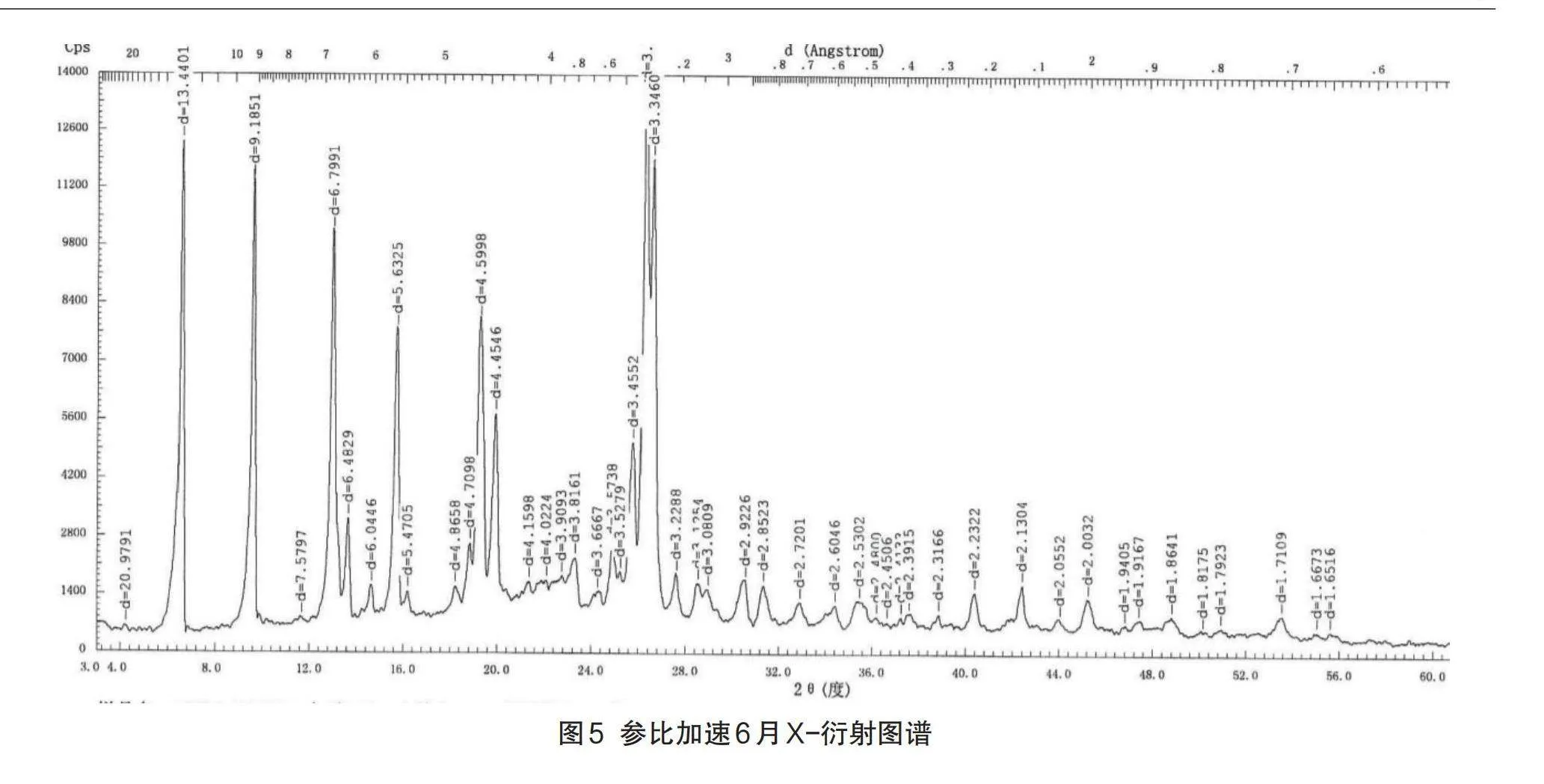
采用X射线粉末衍射法(XRD),对原料药、自制中试制剂、参比制剂进行了晶型对比分析,以判断原料药、自制制剂与原研制剂所使用的原料药晶型是否一致,及制备过程是否对原料晶型产生影响。
1.6含量测定方法
1.6.1色谱条件
试验使用色谱柱:Inertsil ODS C18柱(规格为0.46cm×25cm,5μm);测试波长:360nm;使用流动相:醋酸铵(8.5g)硫酸铜(1.25g)L-异亮氨酸(1.3g)溶液(加水1000ml溶解)-甲醇(70%:30%);流速:每分钟0.8ml;进样体积:25μl;柱温:45℃。
1.6.2溶液的制备
对照品溶液:精密称取约20mg的左氧氟沙星对照品(按C18H20FN3O4计),置于100mL容量瓶中,加入盐酸(0.1mol/L)溶液通过超声处理溶解后稀释定容,摇匀即可得到对照品溶液(浓度约为0.2mg/mL)。供试品溶液:取左氧氟沙星药片10片,经过研磨,精密称取左氧氟沙星约20mg(按C18H20FN3O4计),放入100ml容量瓶中,加入盐酸(0.1mol/L)溶液通过超声处理溶解后稀释定容,摇均匀后过滤即可得到样品溶液。
系统适用性溶液:称取氧氟沙星对照品约20mg,加入盐酸(0.1mol/L)溶液通过超声处理溶解后定量稀释,摇均匀后即可得到系统适用性溶液(浓度约为0.4mg/mL)。
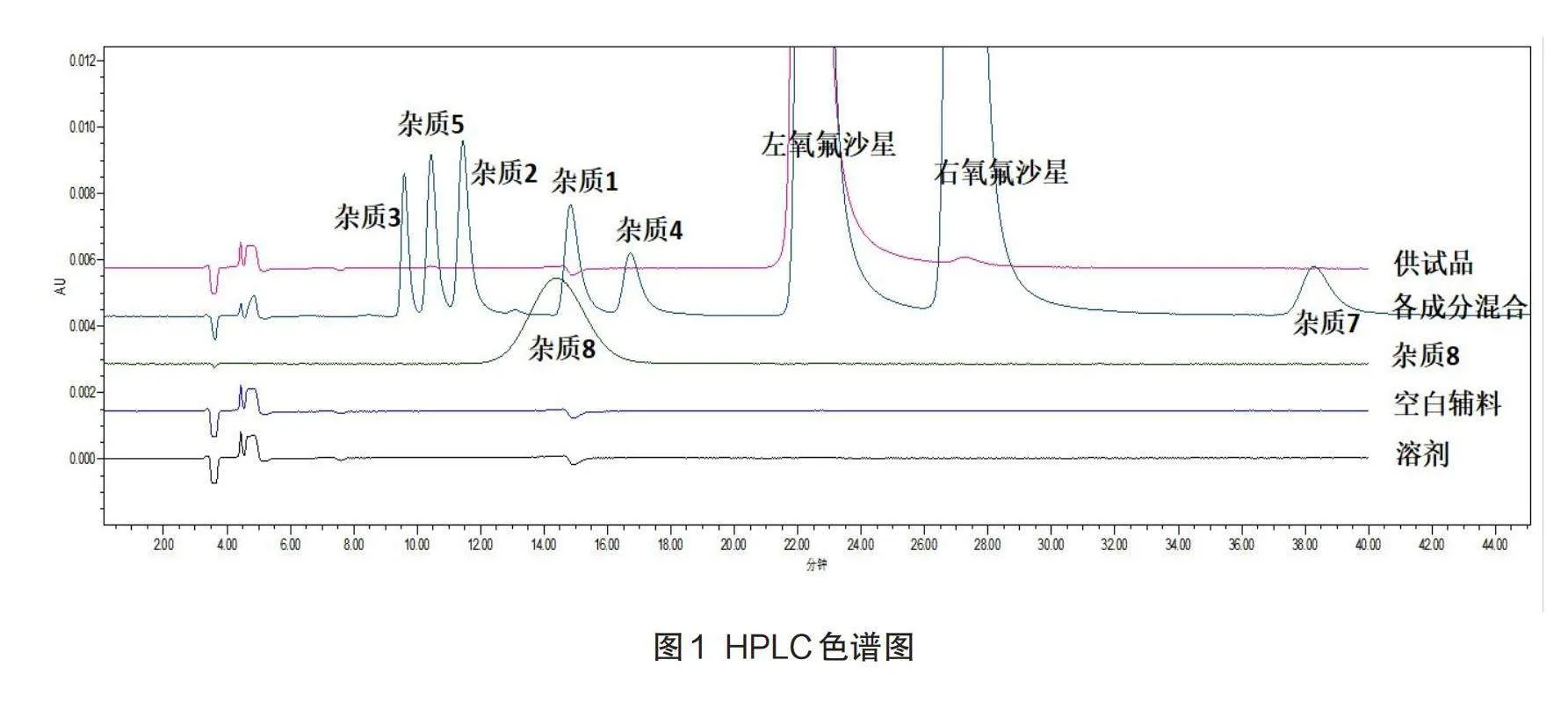
各杂质成分混合溶液:将约10mg氧氟沙星对照品,放入10ml的容器中,使用流动相进行振摇稀释溶解后,定容,摇均匀。分别将杂质1、2、3、4、5和7的储备液(约0.5mg/ml)各0.1ml置于10ml容量瓶中,然后将4.0ml的氧氟沙星溶液也放入同一容器,再使用盐酸(0.1mol/L)溶液进行稀释定量,摇匀即可得到各杂质成分混合溶液。
空白辅料溶液:称取左氧氟沙星包衣片各种辅料,参照供试品溶液制备方法按处方的比例进行制备,即得。
1.6.3含量方法学验证
专属性考察:使用高效液相色谱仪,分别取上述配制的各溶液25μl进行测试,色谱图见图1。结果表明溶剂、空白辅料及各杂质均不干扰左氧氟沙星的含量测定,对照品和供试品在同一位置出现色谱峰,该检测方法专属性良好。
线性关系考察:分别精密量取1.0ml、1.6ml、2.0ml、2.4ml、3.0ml线性储备液,各放入20ml量瓶中,使用盐酸(0.1mol/L)水溶液快速稀释至刻度,充分振匀。分别取各溶液25μl,使用HPLC法,以HPLC色谱图峰面积对线性溶液浓度作图,计算左氧氟沙星含量测定的线性回归方程和相关系数(r)。结果表明在98.74μg/ml~296.21μg/ml浓度范围内,左氧氟沙星色谱峰的面积与其浓度具有良好的线性关系,其回归方程为A=23449.6667C-41306.0897(r=1.0000)。
重复性试验:分别取25μl对照和供试品溶液,使用HPLC法,连续进行6次实验,结果显示,采用外标法计算,可以得出供试品中左氧氟沙星含量(按C18H20FN3O4计)的RSD值为0.3%(n=6),表明该方法重复性良好。
精密度试验:由另一人员于另一时间更换仪器对同一供试品测定,测定方法同重复性,计算左氧氟沙星含量的RSD为0.4%(n=12),表明本检测方法精密度良好。
回收率试验:将3份(约13mg)的空白辅料与约32mg、40mg、48mg左氧氟沙星(按C18H20FN3O4计)工作对照品混合放入100ml容器中,再添加适当的盐酸(0.1mol/L)水溶液,进行超声处理约2min,然后将其冷却到室温,再将盐酸(0.1mol/L)水溶液加入,稀释至所需浓度,振匀后过滤;从续滤液中取5.0ml放入10ml容器中,再将盐酸(0.1mol/L)水溶液加入,稀释至所需浓度,振匀(平行配制3份),得到9份样品,用于回收率试验测试。使用HPLC法,从每个样品中抽取25μl进行测试,按外标法计算,各样品中左氧氟沙星(按C18H20FN3O4计)的回收率平均为100.6%,RSD为0.3%(n=9)。
稳定性实验:在室温条件下,使用HPLC法,分别于0h、2h、4h、6h、8h、10h、12h、16h、20h和24h时抽取25μl供试和对照品溶液进行测试。经过24小时测试的色谱图结果显示,对照品和供试品溶液RSD均不大于2.0%,这表明它的稳定性24小时内良好。
1.7加速稳定性试验考察
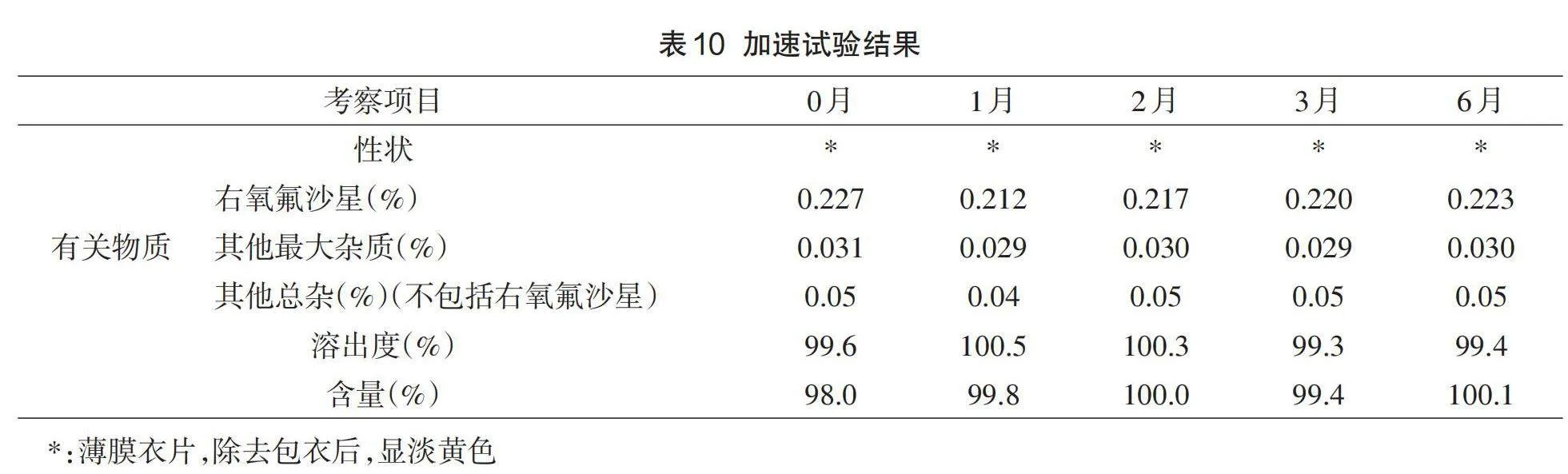
为了考察处方工艺设计的合理性,将自制的左氧氟沙星薄膜包衣片进行温度为40℃±2℃和湿度为75%±5%的加速稳定性试验,将自研左氧氟沙星薄膜衣片进行铝塑包装,放入加速稳定性试验箱内,分别于0月、1月、2月、3月、6月取样,检测左氧氟沙星片的性状、溶出度、含量、有关物质。
2结果与讨论
2.1原研制剂解析
2.1.1原研制剂处方
根据第一三共制药公司在日本上市的左氧氟沙星片IF文件[10],该药物的片芯处方由微晶纤维素、羧甲纤维素、硬脂富马酸钠和羟丙纤维素组成;包衣层处方为羟丙甲纤维素、氧化铁黄、二氧化钛、滑石粉、氧化铁红、聚乙二醇6000、巴西棕榈蜡。以上辅料均为片剂常规辅料,各辅料性质稳定良好。
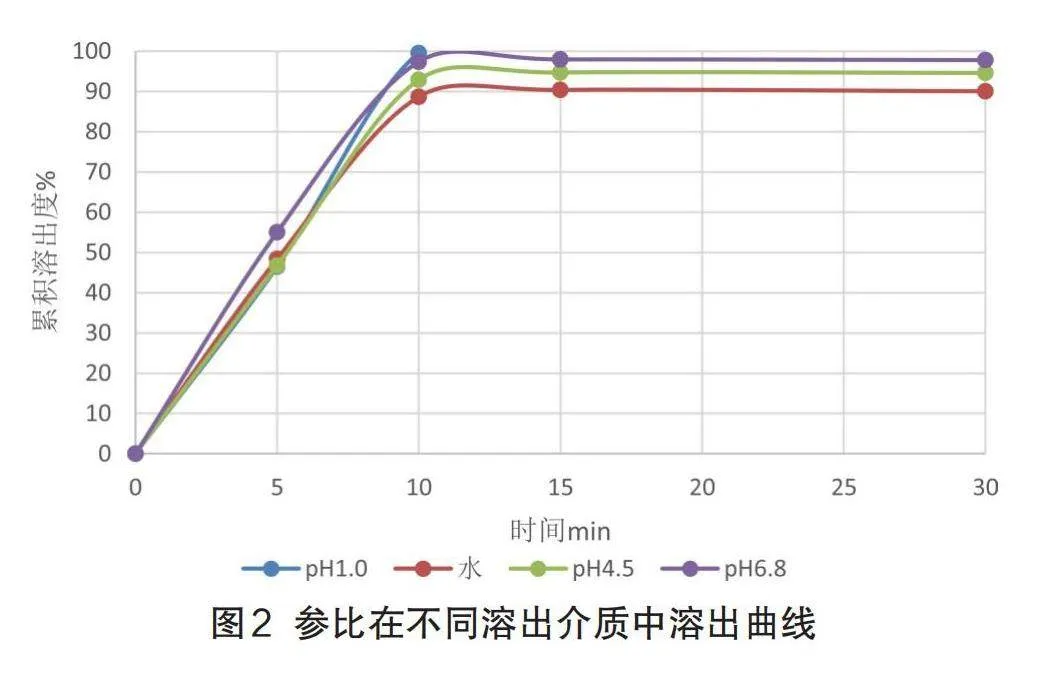
2.1.2原研制剂溶出考察
按1.4项下溶出度检测方法,分别于5min,10min,15min,30min,45min时取样考察原研制剂溶出,结果如图2,原研制剂在磷酸盐缓冲(pH6.8)溶液、盐酸(pH1.0)溶液以及醋酸盐缓冲(pH4.5)溶液和水四个溶出介质中都可以15min达到85%以上的溶出度,为快速溶出。
2.2左氧氟沙星薄膜衣片处方筛选与优化分析
2.2.1润湿剂乙醇浓度的筛选分析
润湿剂不同浓度试验考察结果见表5。经对比用60%、50%、40%的乙醇溶液作为粘合剂配制溶剂,制粒过程顺利,解决了诱发API粘性的风险,三个方案无明显差别,暂定50%的乙醇作为粘合剂溶剂,进行后续处方研究。
2.2.21MGmwdzq3eQuGl04RtfMuw==崩解剂和填充剂的筛选分析
崩解剂和填充剂不同比例实验结果显示:微晶纤维素比例过高时软材不易过筛制备湿颗粒,三批处方干颗粒均易于压片,片面光滑,脆碎度均小于1.0%;三批片剂崩解时间均不超过5min,溶出方面无差异。但微晶纤维素与羧甲纤维素钙比例为8:2时,溶出试验底部堆积较多,考虑到生产放大风险,因此初步选用微晶纤维素和羧甲纤维素钙比例为7:3。
2.2.3粘合剂的筛选分析
经过不同粘合剂用量处方压制药片的硬度、脆碎度、溶出度进行考察表明:粘合剂浓度1.5%脆碎度明显较高,其他方案无明显差异,2.5%~7%浓度可作为本品粘合剂用量的设计空间,结果见表6。
2.2.4润滑剂用量筛选分析
硬脂富马酸钠是一种具有良好润滑性的聚结物,它呈白色或类似白色粉末状[11],具有较强的润滑效果,可以显著降低物料与设备之间的摩擦力,同时也不会影响溶出。经过对不同硬脂富马酸钠加入量压制药片的硬度、脆碎度、溶出度进行考察可知,增加硬脂富马酸钠的添加量不会降低溶出,鉴于本品在压片过程中易发生黏冲现象,所以选用较高添加量4.2%(27mg/片)的硬脂富马酸钠。考察结果如下:
2.2.5包衣厚度筛选分析
对包衣前后溶出变化以及体外溶出与不同包衣厚度的关系的考察表明:包衣前后的溶出无明显变化,不同包衣厚度对药品的体外溶出行为都没有明显的影响,且与原研药品的溶出一致,因此确定包衣增重范围选择3%~5%。
2.2.6原料药晶型研究分析
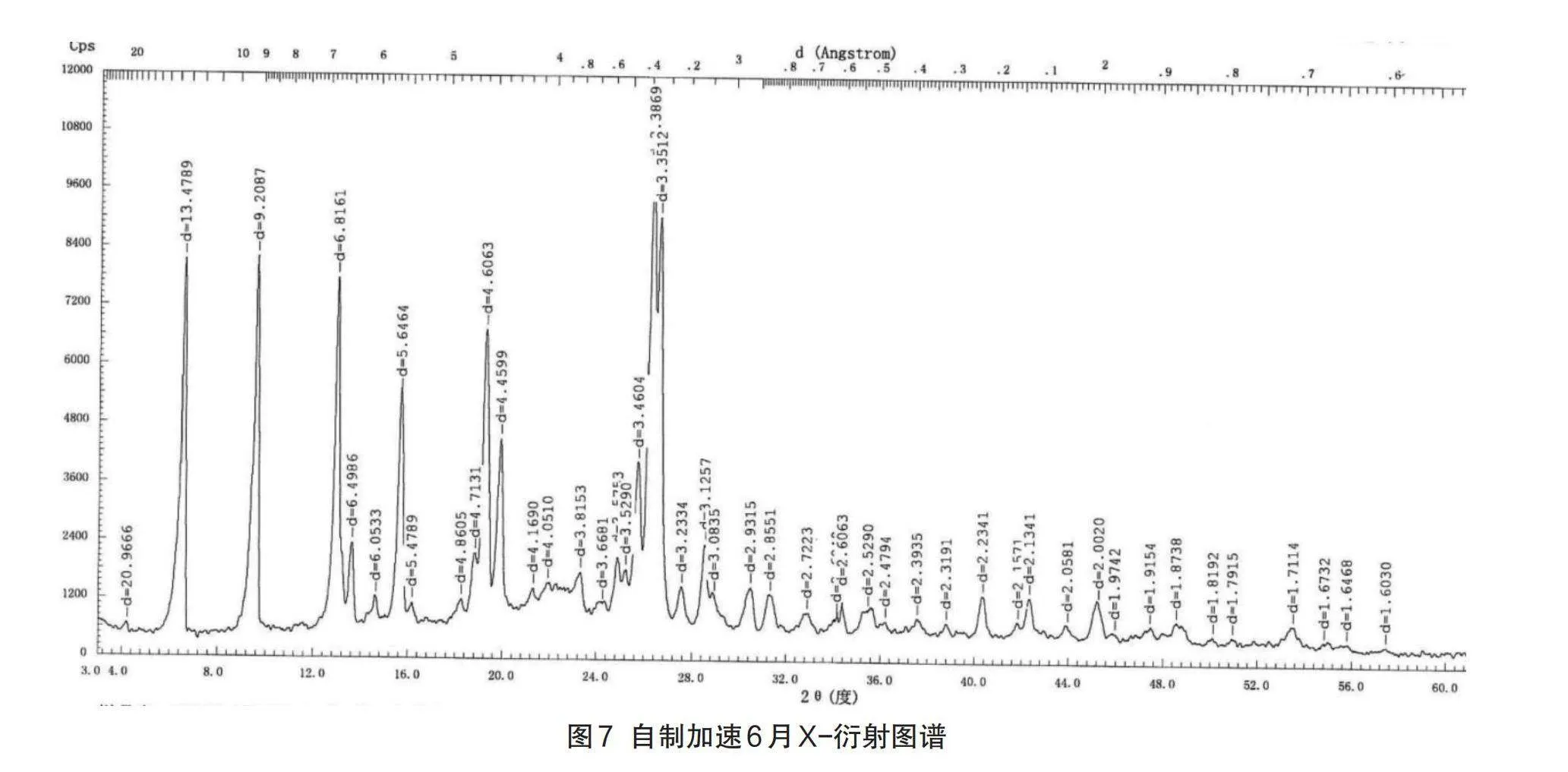
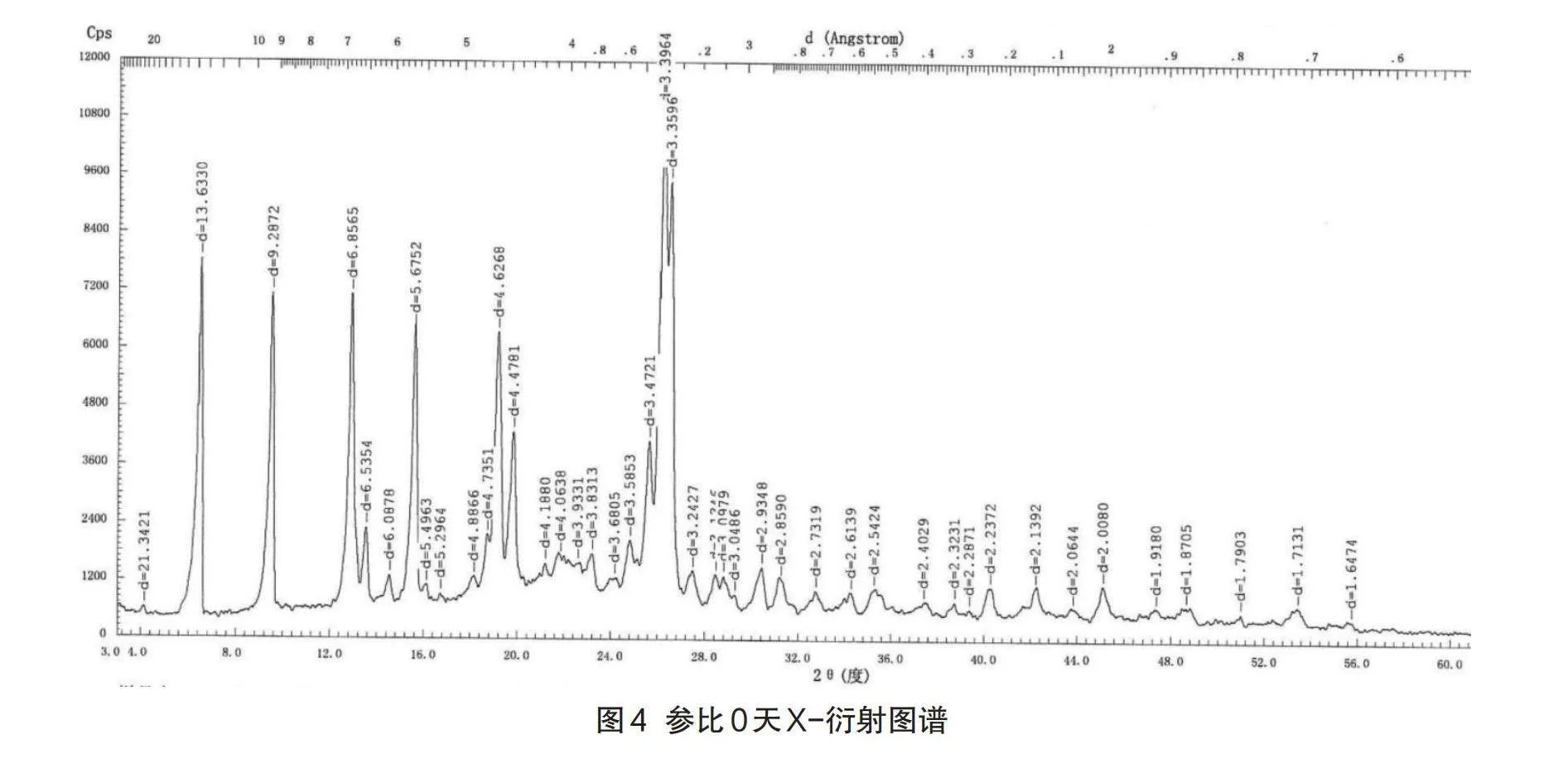
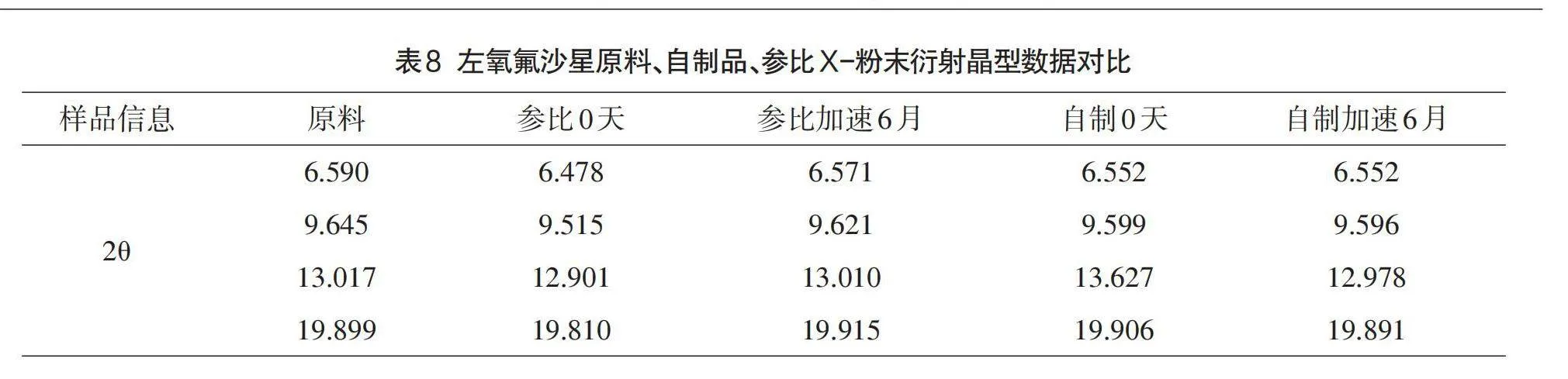
由于不同的晶形结构,其对应的生物、化学、物理特征也会大相径庭,在粉体学性质、外观、熔点、溶解度、溶出速率、化学稳定性、治疗作用等方面可能会有显著不同[12],因此,其对于药物的稳定性、生物利用度以及治疗作用的影响也是极其重要的[13],这种影响在口服固体制剂中表现得最为突出。左氧氟沙星原料药具有多种晶型,其半分子水的结晶物为临床广泛应用的稳定晶型[14],性状为针状结晶性粉末。X射线粉末衍射法(XRD)结果显示:原料药、原研制剂及自制制剂晶型一致,6月加速考察后自制品和原研制剂晶型均未发生变化。各代表批次粉末衍射数据表8、图谱见图3-图7。
2.3左氧氟沙星片的质量评价
2.3.1含量测定
取三批自研样品和三批原研样品,按1.6项下供试品溶液制备方法制备及含量测定方法测定含量,结果三批自研和原研样品的含量分别为98.38%、98.61%、97.99%,99.81%、100.69%、98.74%,均在95%~105%内,符合规定。
2.3.2溶出度测定及溶出曲线的相似性对比研究
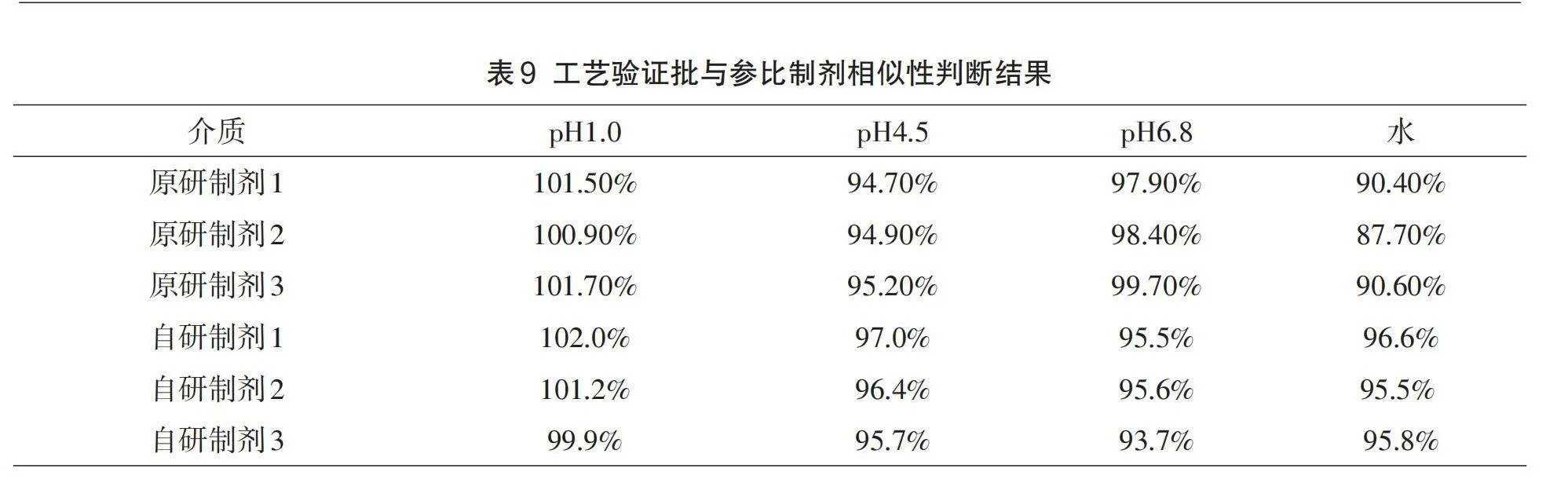
溶出度是口服固体制剂衡量自研制剂和原研制剂质量一致性的重要指标,它能够有效地提高临床疗效的一致性,可用来豁免部分仿制药制剂开发过程中的生物等效性试验。根据《人体生物等效性豁免指导原则》《普通口服固体制剂溶出度试验》等国内外指导原则,采用相似因子(f2)法判断相似性,如果参比产品和仿制产品均可以在15分钟内达到85%或更高的溶出度值,可表明它们的溶出曲线一致(无需再使用相似因子法来确定其相似性)[15]。
取自研左氧氟沙星片和第一三共公司产品,按照1.4项下的溶出介质和溶出度测试方法进行检测,在15min时间点抽取测试溶液10ml,过滤后使用紫外-可见分光光度法进行分析[16]。经计算自制左氧氟沙星片和第一三共公司产品在所选择的4种溶出介质中15min时间点的溶出度值均可以大于85%,这表明它们的体外溶出行为相似,预示自研制剂和原研制剂具有生物等效性,为豁免体内生物等效性提供前提条件。
2.4加速稳定性试验考察分析
加速试验结果见表10。左氧氟沙星薄膜包衣片加速稳定性试验结果表明,在加速条件下放置6个月,右氧氟沙星、其他最大杂质和总杂均未出现增加趋势,性状、含量和溶出度均无明显变化,质量符合要求。
3结论
本研究通过对原研制剂的解析、自研制剂制备的处方工艺的摸索、质量评价和加速稳定性试验,初步确定了自制左氧氟沙星薄膜包衣片制备的处方工艺,自制制剂和原研制剂具有相同的晶型,相近的含量,相似的体外溶出行为,加速稳定性期间各项质量指标变化趋势一致。综上所述,自研制剂和原研制剂质量具有一致性,预示自研制剂和原研制剂具有生物等效性。为进一步产业化左氧氟沙星片提供科学参考依据。
参考文献:
[1]韦志成,秦天娥,蓝海江.左氧氟沙星治疗急性细菌性感染性腹泻的临床效果[J].中国社区医师,2023,39(12):56-58.
[2]曾金萍,徐海胜,张腾飞.左氧氟沙星注射液辅助治疗在肺结核患者中的应用效果观察[J].现代医学与健康研究,2024,8(2):7-9.
[3]归莱,张桂芬,王建.某院117例左氧氟沙星所致药物不良反应的流行病学分析[J].抗感染药学,2023,20(9):908-913.
[4]董菊红,刘宁,陈雪薇.我国仿制药质量一致性评价进展分析与建议[J].中国处方药,2023,20(12):1-5.
[5]刘瑜,裴育.一致性评价政策解读及仿制药与原研药差距分析[J].基层医学论坛,2022,26(31):135-138.
[6]罗国良,徐秀卉,宋远征,等.企业开展仿制药一致性评价的常见问题与建议[J].医药导报,2021,40(12):1780-1784.
[7]刘增平,黄洁.盐酸左氧氟沙星片处方优化研究[J].中国药业,2015,24(14):40-41.
[8]徐小乐,王燕,曾庆福.苎麻骨纤维素提取及微晶纤维素制备工艺研究[J].武汉纺织大学学报,2013,26(3):1-5.
[9]张润东.功能性羧甲基纤维素/壳聚糖复合材料的制备及性能的研究[D].武汉纺织大学,2019.
[10]医療用医薬品の添付文書情報,レボフロキサシン錠(第一三共エスファ株式会社)-IF利用の手引きの概要[EB/OL].[2023-10-31].https://www.info.pmda.go.jp/go/pack/6241013C2032_1_11/?view=frame&style=XML&la-ng=ja.
[11]国家药典委员会.中华人民共和国药典四部[M].北京:中国医药科技出版社,2020.
[12]周丰涛,彭亚运,蔡挺.多晶型对药物固体理化性质影响的研究进展[J].中南药学,2023,21(11):2984-2990.
[13]杨文智,宁黎丽,许真玉.晶型研究在口服固体制剂仿制药药学研发中的几点关注[J].中国药学杂志,2022,57
(15):1302-1304.
[14]舒理建,沈晓峰,杨平爱,等.左氧氟沙星半水合物的制备工艺[J].中国医药工业杂志,2020,51(6):713-715.
[15]申际丽,马坤,龚青,等.速释口服固体制剂人体生物等效性试验豁免药学研究的要求[J].药物评价研究,2023,46(3):493-500.
[16]郑红,宋立倩,房东阳.盐酸左氧氟沙星缓释片的制备及药物释放影响因素的考察[J].中南药学,2021,19(8):1575-1580.
The Quality and Preparation of Levofloxacin Film-Coated Tablets
ZHANG Zhen1,CHENYajun1*,GONG Jiangxue2,ZHANG Lan2
(1.School of Medicine,Wuhan University of Science and Technology,Wuhan Hubei 430065,China;
2.Hubei Duorui Pharmaceutical Co.,Ltd.,Jingzhou Hubei 434300,China)
Abstract:The consistency of the quality between the self-crafted and original preparations can ensure the clinical effect of the two prepara-tions to agreater extent.Therefore,the particle forming,fragility,and in vitro dissolution was used as the evaluation indicators,the prescrip-tion of self-developed formulations by screened and optimized experiments of the types of wetting agents,fillers and disintegrants,lubri-cants,adhesives,and coating powders.The optimum prescription of levofloxacin film-coated tablet contained Levofloxacin 512.5mg,micro-crystalline cellulose 70mg,calcium carboxymethyl cellulose 30mg,hydroxypropyl cellulose 3.75mg and sodium stearate 27mg.Coating ma-terial 3-5%was the optimal weight of levofloxacin tablets.Conduct quality inspection on the film coated tablets prepared by the selected pre-scription and compare them with the original research formulation,the self-developed drug and reference listed drug,when dissolved in four media-phosphate buffer solution(pH 6.8),hydrochloric acid(pH 1.0),acetate buffer solution(pH 4.5),and water-all had dissolution rates higher than 85%for 15 minutes,suggesting asimilar dissolution behavior.Same crystal form and content;there was no significant change in the sample control indicators after 6 months of accelerated tested compared to 0 days.It is proved that the formulation and technology of self-developed levofloxacin tablets are stable and feasible,and the quality was consistent with the original preparation.
Keywords:Levofloxacin;Dissolution rate;Stability;Quality consistency
(责任编辑:李强)
