
LA-SA\\MMA相变微胶囊的制备及其性能研究
2024-11-01 00:00李想许宛平柯贵珍
武汉纺织大学学报 2024年5期


摘要:通过原位聚合法,制备了以甲基丙烯酸甲酯为壁材,月桂酸、硬脂酸共熔物为芯材的相变微胶囊LA-SA\MMA相变微胶囊。利用SEM扫描电镜、差示扫描量热仪、马尔文粒径分析仪、傅里叶红外光谱分析仪和TG热重分析仪观察并检测不同芯壁比的微胶囊,结果显示:芯壁比为4:5微胶囊呈圆球状、表面光滑平整,但是略微存在团聚粘着现象。囊壁对芯材起到了一定的保护作用,有效地提高微胶囊的热稳定性。微胶囊耐久性十分优异,可以多次重复使用。然后,使用干法涂层技术把不同质量分数的相变微胶囊涂覆到SMS织物上,产生相变温控织物。通过热成像,讨论经过相变微胶囊涂层整理后的织物,其蓄热控温性能是否受到影响。结果显示:经过涂层整理后的织物,其降温速率下降变慢,能够应对外界环境温度的变化。
关键词:甲基丙烯酸甲酯;月桂酸;硬脂酸;相变微胶囊;原位聚合法
中图分类号:TS102.1文献标志码:A文章编号:2095-414X(2024)05-0060-07
0引言
相变材料是根据在一定温度范围内的环境温度的变化实现智能温度调节效果,吸收外部热量或释放其中存储的热量的材料[1]。而初始相变材料存在诸如泄漏、相分离、体积膨胀、热稳定性差和使用期间的腐蚀性等问题,这限制了它们的使用[3]。在20世纪后期,美国公司制备出来可以交换热量的功能性微胶囊,并将这类胶囊通过纺织品的后整理附着,使纺织品具有调温能力。相变微胶囊利用物理或化学的方式,ZplKrODYFtwKjVK/qb0E0Q==将囊壁材料聚合包覆住具有特定相变温度的相变芯材料[4]。微胶囊技术的最终成品是复合相变材料,该种材料的核-壳结构应十分稳定[5]。所述核-壳结构的粒径一般在0.1-100μm的范围内,囊壁厚度为一般不超过10μm,太厚会导致传热受阻,也不能低于0.1μm,过薄会使得胶囊的强度和耐磨性能变差[6]。相变微胶囊材料可通过吸收和释放热量来调节和控制材料周围环境的温度。相变微胶囊可以根据其施加环境的温度条件选择具有最佳相变温度的芯材料[7]。
就微胶囊技术本身而言[9],目前使用的大多数方法是原位聚合。采用的壁材以三聚氰胺一甲醛树脂及脲醛树脂为主,这种壁材原料价廉易得,具有良好的机械强度和广泛的应用范围[10]。
周宇飞、袁一鸣[11]等制备了用三聚氰胺-甲醛-尿素树脂包覆石蜡囊芯的微胶囊。采用了原位聚合法制备出的微胶囊粒径均匀且外观良好,石墨烯量子点的加入有助于提高微胶囊粒径的均匀性,同时复合石墨烯量子点及纳米铝的微胶囊导热系数提高了254.55%,达到0.78W/(m·k),包覆率提高至92.65%。周龙祥、王保明[12]等采用溶胶-凝胶法制备了用二氧化钛壳材包覆石蜡芯材的微胶囊、乳化剂是十二烷基硫酸钠。微胶囊呈圆球形,粒径在80~90μm之间,微胶囊包覆率达到了69.36%,且微胶囊具有良好的热性能和热稳定性能。
总之,在已有的研究[13]当中,相变微胶囊壁材多以脲醛树脂、酚醛树脂等合成高分子材料为主,并且很多研究人员会对壁材进行改性,在其中加入各种纳米颗粒或制成复合壁材,以达到相变微胶囊的增强、吸磁、调湿等多种综合性能[15]。但是由于这些壁材含有甲醛,对于环境的污染和健康的危害限制了其发展[17]。芯材大部分仍以石蜡、十八烷为主,但是十八烷的价格比较昂贵。近年对醇类的研究逐渐增多,其中聚乙二醇因其良好的相容性、价廉易得、体积变化小是研究的热点[18]。由于脂肪酸类的相变温度适宜,对于癸酸、月桂酸、棕榈酸和十八烷酸等脂肪酸类相变芯材的研究也开始升温[20]。
本文以月桂酸、硬脂酸共熔物为芯材,可以制得合适相变温度的微胶囊。而以甲基丙烯酸甲酯为壁材,所得到的产品纯度高,无需做后续处理,而且甲基丙烯酸甲酯的耐冲击强度和低温性能良好,引发聚合反应条件简单易行。采用原位聚合法合成LA-SA/MMA,利用SEM扫描电镜、DSC相变潜热、粒径分析、红外光谱分析、TG热重分析和对不同芯壁比的相变微胶囊进行表征。然后,采用干法涂层工艺将不同浓度相变微胶囊涂覆到SMS织物上,形成相变调温织物。通过热成像,探讨相变微胶囊涂层整理对织物蓄热控温能力的影响。
1实验部分
1.1实验原料及设备
硬脂酸、月桂酸、Tween-60、甲基丙烯酸甲酯,国药集团;聚乙烯醇、过硫酸铵,西陇化工股份有限公司;海立柴林粘合剂,上海申致化工科技有限公司。
仪器有HH-4型数显恒温水浴锅(常州国字仪器制造有限公司);DF-101S型集热式恒温加热磁力搅拌器(上海东玺制冷仪器设备有限公司);JM-A10002型电子天平(诸暨市超泽衡器设备有限公司);FJ300-SH型数显高速分散均质机(上海标本模型厂制造);SHZ-DⅢ型循环水真空泵(巩义市予华仪器有限责任公司);DHG-9030A型电热恒温鼓风干燥箱(上海索普仪器有限公司);PS-20A型精准数控超声波清洗机(洁康科技有限公司);UPH-Ⅰ-5/10/20t型UPH标准型超纯水器(四川优普超纯科技有限公司)。
1.2实验原理
1.2.1 LA-SA共熔物
脂肪酸类属于应用广泛的相变材料之一。脂肪酸有很多优点:过冷度比较小、价格便宜,廉价易得、熔化和结晶的可逆性也非常好。此外,脂肪酸间具有很好的混合性能,并且可以混合多种脂肪酸以获得二元甚至多组分的低共熔混合物。以一定比例混合它们会产生最低熔点,这个被称为最低共熔点。共熔以后形成的混合物被称为低共熔混合物。降低温度,该混合物会逐渐冷却至凝固。这种共熔物性质与原来各组份相比,发生了很多变化,但是低共熔混合物的性质是最不易发生改变的,低共熔混合物的相变温度就是低共熔点,也是相变点。以这种方式,可以获得调温纺织品的合适熔化温度。
1.2.2甲基丙烯酸甲酯聚合机理
相变微胶囊的囊壁材料是由甲基丙烯酸甲酯在加热和过硫酸铵的引发下,发生本体聚合形成聚甲基丙烯酸甲酯。在游离基聚合反应中,引发剂是产生活性中心的物质。根据引发剂的溶解性,大体可分为油溶性引发剂和水溶性引发剂两大类。按其化学组成可分为偶氮类、有机\无机过氧类引发剂和氧化还原引发剂。乳液聚合一般采用水溶性引发剂,本实验采用的过硫酸铵是无机过氧化物,属于水溶性引发剂。过硫酸根离子均裂成2个自由基引发剂,引发甲基丙烯酸甲酯发生链增长反应,最后的歧化终止。
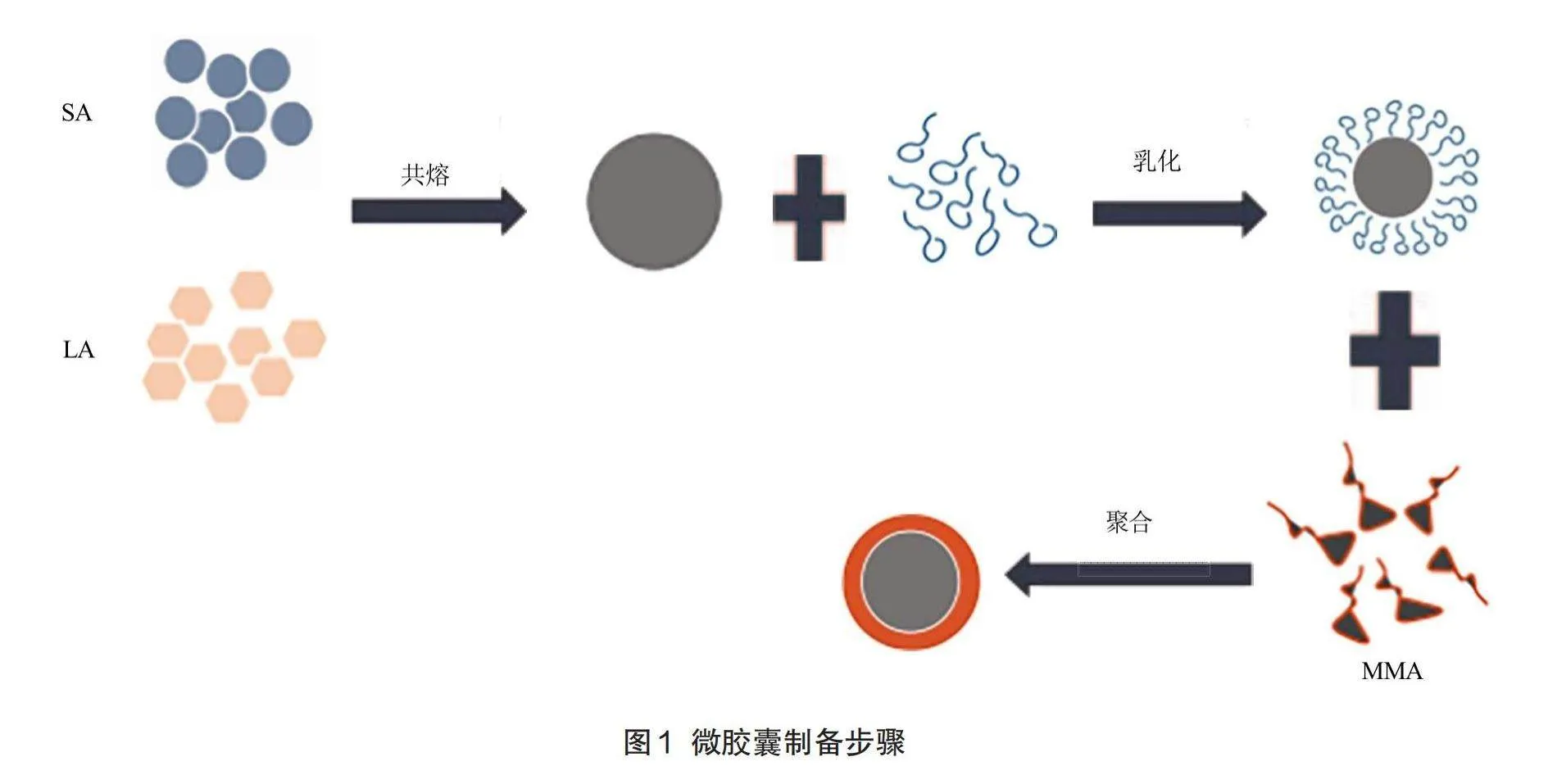
1.3相变微胶囊的制备
1.3.1 LA-SA共熔物的配制
用天平分别称取摩尔组成为LA:SA=6.5:1,即月桂酸为6.57g硬脂酸1.43g,将其混合,并用玻璃棒不断搅拌。将混合后的LA-SA颗粒放入烘箱中,烘箱温度设置为80℃,烘燥时间2h。两小时后取出共熔物,立即放入超声波中超声,水浴温度为60℃,超声时间5min。最后,将共熔物室温冷却,密封保存。
1.3.2共熔物的乳化
将水浴锅的温度设置为70℃,称取8g共熔物于三颈烧瓶中,将三颈烧瓶固定在水浴锅中,加热至共熔物固体全部融化。加入0.5g乳化剂吐温60,稳定剂聚乙烯醇0.4g,搅拌转子的速度设置为600r/min。将纯水机制备的去离子水加热到85℃,于滴定管中匀速滴入到乳液中,整个滴定过程时间控制在20~30min以内。将乳化仪的速度逐渐上升到5000r/min,将滴加完毕后的溶液乳化分散5min,得到LA-SA乳液。
1.3.3制备微胶囊浊液
将乳液倒入三颈烧瓶中并搅拌5min。停止搅拌后,迅速加入1.4g过硫酸铵,同时滴加甲基丙烯酸甲酯4~14g(每增加2g为一组),滴加速度控制在1~3g/min以内。滴毕后,立刻将三颈烧瓶移至温度为85℃的水浴锅中,并将搅拌转子速度提高到800r/min,加热搅拌反应2h。
1.3.4相变微胶囊粉末制备
将微胶囊浊液移至烧杯中,在干燥室温下静置5h,浊液完全冷却沉淀后,取出浮在上层的未被包覆的LA-SA共熔物。将剩余的浊液倒入垫有滤纸的漏斗中,漏斗装置在抽滤瓶上,用循环水式真空泵抽滤机抽滤,抽干后得到滤饼。用镊子将滤饼取出,充分溶解于65℃的去离子水中,再次倒入漏斗进行抽吸。取出第二次抽滤后的滤饼,用少量常温的去离子水洗净。将洗净的滤饼放入培养皿中,放入恒温烘燥箱中,在40℃中烘干滤饼。烘燥结束后,将滤饼等分。在研磨皿中充分研磨。实验步骤如图1。
1.3.5相变微胶囊/SMS织物
在烧杯中加入3%的扩散剂和聚乙烯醇,再倒入粘合剂UDT和一定比例的去离子水。称取一定质量的微胶囊粉末,制备成梯度为10%质量分数从10%到30%相变微胶囊胶液。将微胶囊整理溶液涂覆在SMS布上,并在烘箱中在70℃的温度下干燥2小时。
1.4测试与表征
1.4.1相变微胶囊形貌结构分析
使用Phenom ProX台式扫描电子显微镜(Phenom,荷兰)观察和检测制备的相变微胶囊的表面形态和结构。将相变微胶囊粉末粘附到导电胶上,并在真空喷金后进行真空观察。
1.4.2粒径分析
利用激光粒度仪(Malvern,英国)对所制得的相变微胶囊粒径进行测量分析。将所得的微胶囊粉末中加入一定分散剂与去离子水制得相变微胶囊分散液,装入石英比色皿中对其进行粒径测量。
1.4.3红外光谱分析(FTIR)
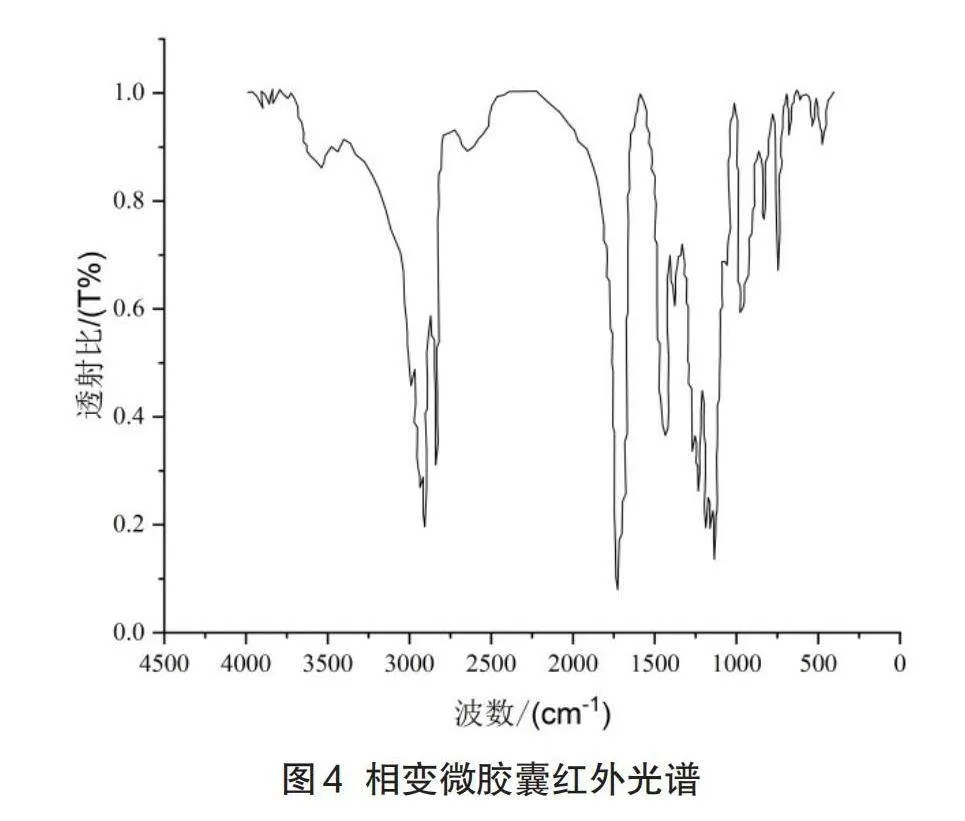
通过FT-IRVERTEX70傅立叶变换红外光谱仪(Bruker,德国)对LA-SA芯材,MMA壁材料和相变微胶囊的化学组成进行分析。傅里叶红外变换光谱是利用物质中的官能团吸收各种波长的红外光的不同程度来判别。由于物质的分子结构有所差异,根据红外光谱中吸收峰波段的位置,数目和形状,定性地确定物质中的官能团,进而推断出物质分子构成。实验采用KBr压片法,红外光谱显示的波长在400~4000cm-1以内。
1.4.4差示扫描仪测试
通过DSC-204 F1(NETZSCH,德国)差示扫描量热仪对相变囊芯LA-SA、LA-SA\MMA相变微胶囊进行DSC测试。分析得到DSC曲线,得到LA-SA\MMA相变微胶囊的相变温度、相变的热量变化。测试条件:使用氮气作保护气体,流速为20mL/min,每分钟升温10K,起始温度为0℃,终止温度l45gTZl76eD9OTB59QbMjw==为100℃。
对LA-SA\MMA微胶囊进行温度循环实验,一次循环分为两个阶段:第一个阶段是从10℃升温到60℃,第二个阶段是从60℃再降到10℃。
1.4.5热重性能分析
LA-SA相变微胶囊的耐热性试验通过热重分析仪TG209F1(NETZSCH,德国)进行。随着温度或时间的增加,测量坩埚中物质的物质的量。这个测定过程由热重分析仪程序控制。它用于测量相变材料,相变微胶囊壁材料和相变微胶囊的热稳定性。氮气作为环境气体,升温速率10K/min,温度范围为0℃~600℃。
1.4.6相变微胶囊/SMS织物热成像
将涂有不同微胶囊浓度含量的三种涂层织物和未涂覆的普通织物同时在70℃的烘箱中1小时。之后,迅速取出,并且每10秒用FLIR热成像相机测量织物的表面温度,直到织物降至室温,并绘制冷却曲线。
2结果分析与讨论
2.1微胶囊的形貌
图2显示了在三种不同的芯壁比条件下制备出的微胶囊电镜图。芯壁比为1:1的反应体系中没有完全形成微胶囊,并且有破碎的孔洞和边缘,这表明MMA的聚合有很大一部分没有完成,或者是形成的微胶囊壁太薄强度不够,在真空抽滤的过程中,没有形成完整包覆的微胶囊壁破裂。芯壁比为4:5的微胶囊基本上是球形的,并且微胶囊的表面是光滑和平整的,几乎没有棱角或凹痕。芯壁比为2:3的反应体系发生了团聚现象,产生的微胶囊数目很少且破损十分严重。
2.2 PSD粒度分析
由图3可以看出,LA-SA/MMA微胶囊样品粒度分布图呈现出两个明显的区域,微胶囊的直径主要分布在0.4~1μm和6~13.5μm两个区间,经过计算,平均粒径比6μm还要低出0.04μm。
通过粒度大小分布和与扫描电镜图片对比发现,粒径非常小的微胶囊约占33%,其直径一般在0.3~1μm内,这部分微胶囊的平均粒径为0.39μm;在6~1.3.5μm的微胶囊占了很大比重,达到了约67%,这部分的平均粒径为7.99μm。出现这种分布差异和粒径异常,可能是由于微胶囊浊液在石英比色皿中分散不足,导致微胶囊之间的团聚和粘附。
2.3红外光谱分析
在图4中,2953.6cm-1处对应甲基中的碳氢键不对称伸缩振动吸收峰。对应于中碳氢键的2856.1cm-1对称伸缩振动吸收峰,酯基中的羰基伸缩振动吸收峰在1737.3cm-1处,对应甲基中的1453.3cm-1的碳氢不对称变形振动吸收峰,1187.1cm-1、1151.3cm-1处对应酯基中的碳氧单键的伸缩振动吸收峰,这些官能团与PMMA的特征谱带完全相符合,证明微胶囊中壁材料的形成。1691.28cm-1是硬脂酸中羧基上的羰基的特征峰;在3000cm-1时,只有峰出现在了右侧,表明被测物质仅具有饱和的碳氢键。没有不饱和的碳氢键,这与饱和脂肪酸如月桂酸、硬脂酸的结构一致。在1660~1640cm-1之间没有明显的吸收峰,表明没有碳碳双键的特征峰,表明甲基丙烯酸单体完全聚合。制备的微胶囊中含有脂肪酸和聚甲基丙烯酸甲酯的组分,并且微胶囊在干燥过程中没有出现芯材的熔融溢出,这表明甲基丙酸甲酯聚合成了壁材料且脂肪酸被包封以形成微胶囊。
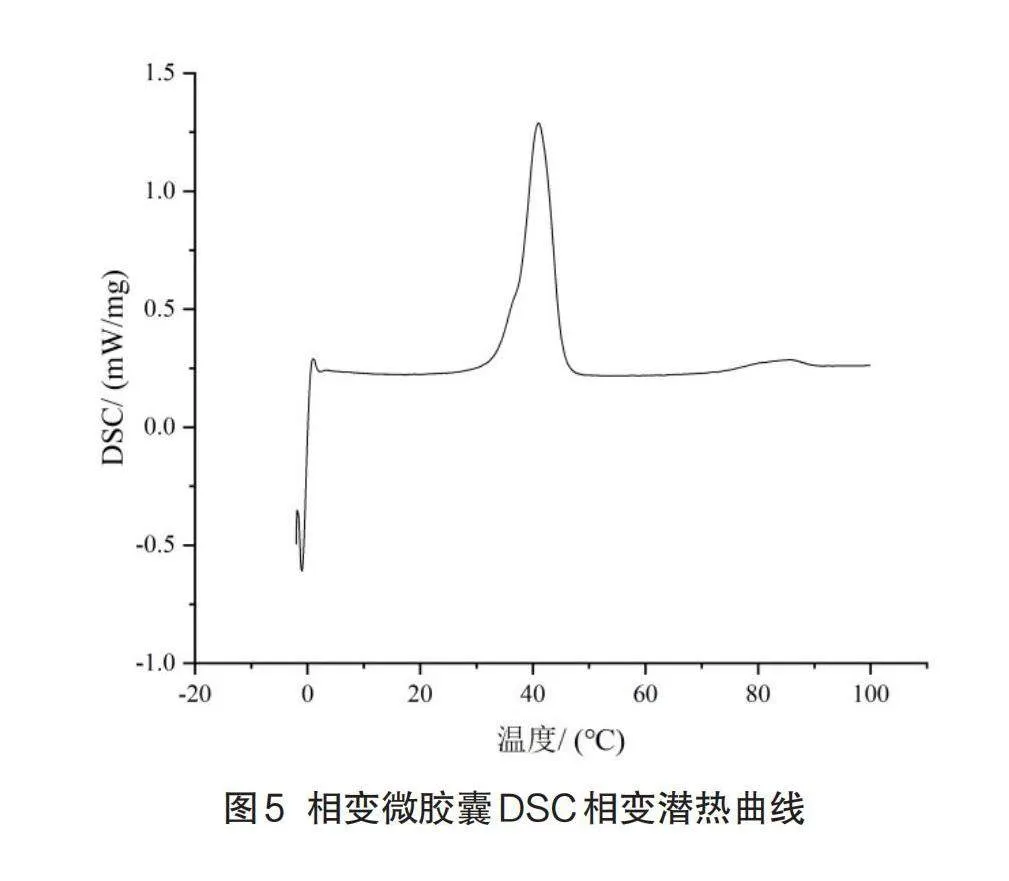
2.4 DSC分析
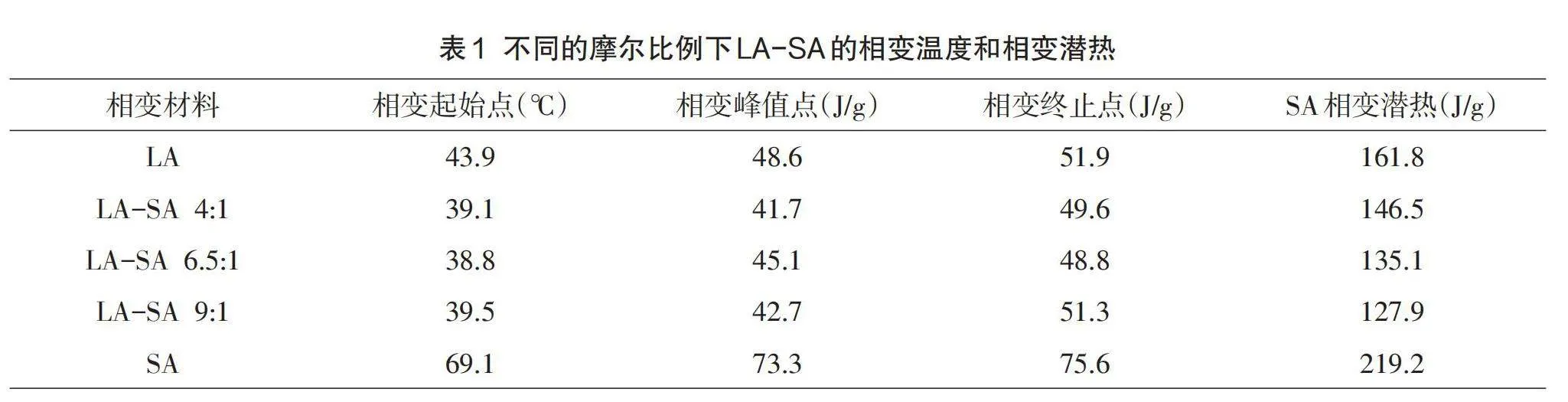
图5显示了相变微胶囊的DSC曲线,小峰则对应脂肪酸共熔物的固-固相变,主峰代表芯材脂肪酸共熔物的固-液相变。起始曲线稳定,峰值起始温度为36.5℃,峰值温度为41.1℃,终止温度为45.5℃,升温相变潜热为28.92J/g。包埋后相变温度、相变起始点、终点都略微减小,但变化不大。
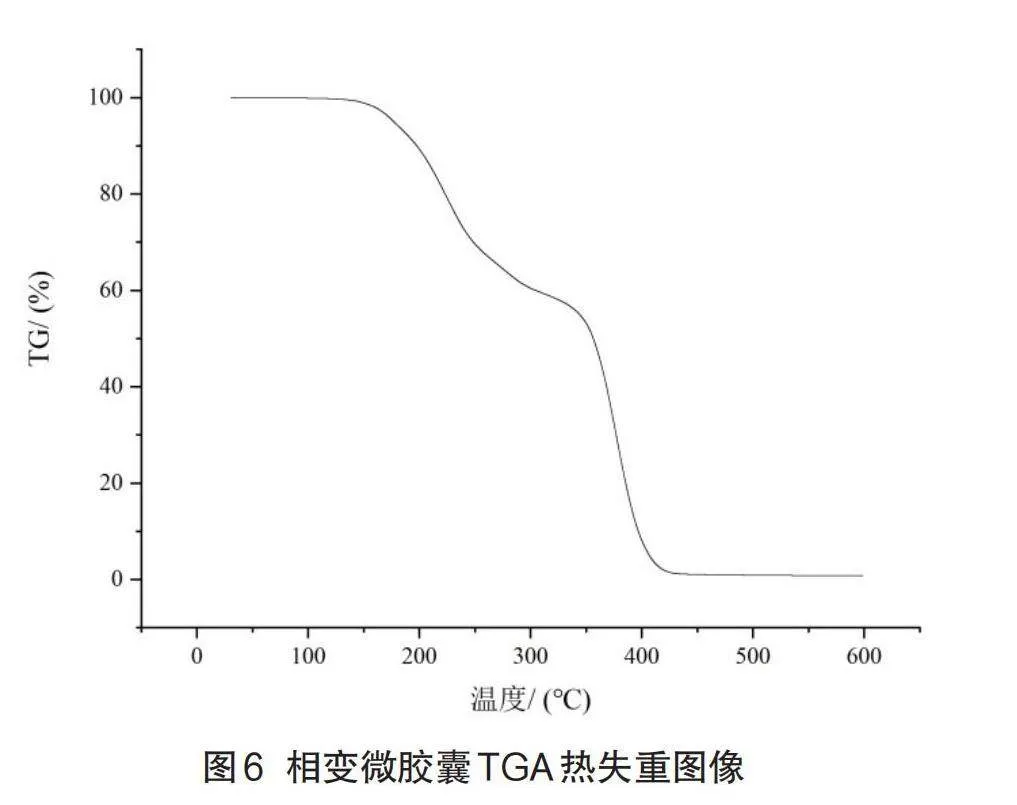
2.5 TGA热重分析
图6为相变微胶囊的热失重曲线,由图可知从138.7℃开始分解到179.2℃,这是第一失重阶段,失重率为6.4%,这个阶段的失重主要是由于芯材脂肪酸共熔物的分解和逸散。温度持续上升,直到320.1℃时,样品质量减少了42%,升温95.5℃直到415.6℃,样品全部分解失重,这个阶段失重曲线相比前一部分的斜率更大更为陡峭,说明失重速率加快。与无囊壁包被的共熔物热失重曲线相比,LA-SA/MMA相变微胶囊的热失重曲线相对平缓、热分解速率有所减小,芯材脂肪酸共熔物的耐热温度与直接使用时有所提高。
这表明PMMA能囊壁对芯材起到了一定的保护作用,有效地提高微胶囊的热稳定性。
2.6相变微胶囊/SM S调温织物热成像
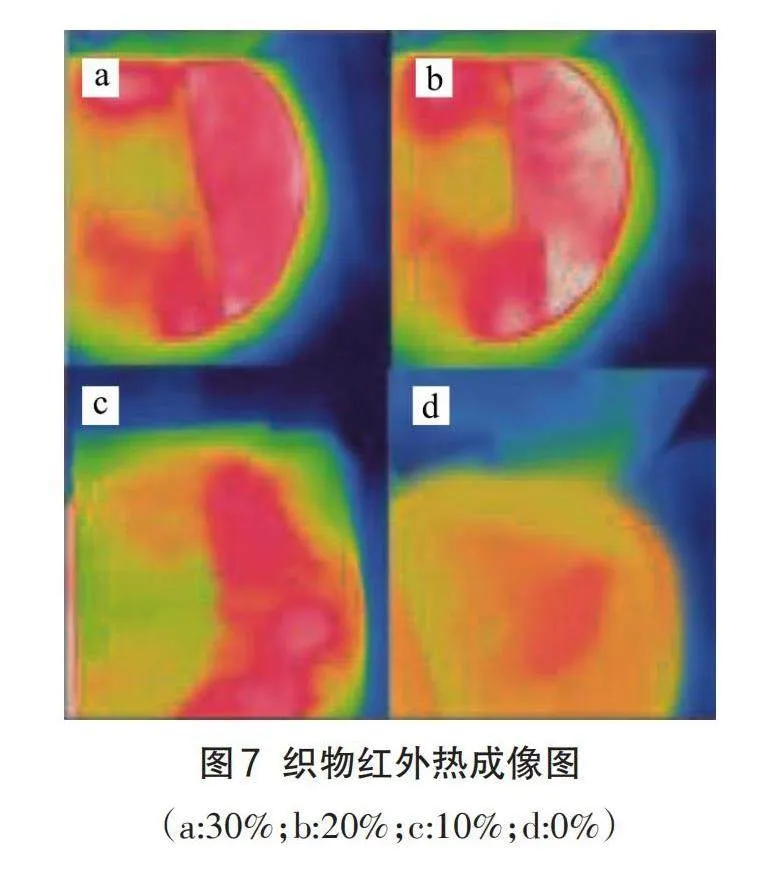
图7显示了织物在加入不同含量的微胶囊后的红外热成像图,可以看出随着涂层中微胶囊的含量增加,织物表面温度明显得到提升。
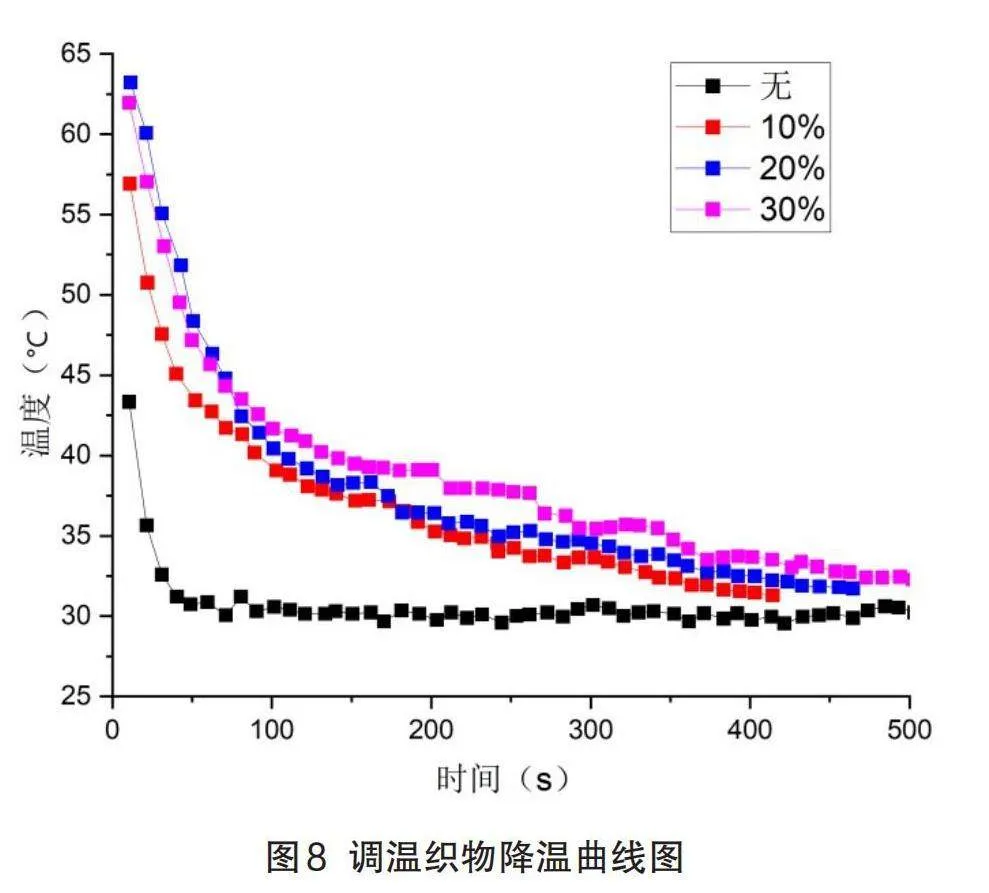
如图8所示,原织物的降温曲线陡峭,但是加入了微胶囊的调温织物,会在降温过程中与外界环境进行热量交换,其降温曲线明显更加平缓,在LA-SA相变芯材的相变温度附近出现拐点,大约在38~42之间,符合DSC测出的数值。随着涂层中微胶囊的含量增加,由0逐渐增加到30%,曲线的斜率减小,即冷却速率逐渐降低。当温度降到室温左右时,调温织物表面的温度始终略高于未经过整理的织物。这是由于温度降低时,LA-SA芯材由液相转变为固相,向外界环境放出热量。
综上所述,经过LA-SA\MMA微胶囊整理后的织物具有更好的储热和温度调节能力。
3结论
本文以甲基丙烯酸甲酯为壁材、月桂酸与硬脂酸共熔物为芯材,利用原位聚合法成功制备出LA-SA/MMA相变微胶囊,并研究了该微胶囊的热性能,得出以下结论:
(1)LA-SA/MMA相变微胶囊的SEM和粒度分析表明,实验制备的相变微胶囊呈球形。表面光滑平整,彼此之间具有粘连团聚的现象,并且分散性一般。包覆良好,可有效防止芯材泄露。相变微胶囊的平均粒径为5.96μm,粒径最大达到13.5μm,最小为0.42μm。
(2)DSC分析表明,在升温时,LA-SA/MMA相变微胶囊起始相变温度为36.5℃;结束相变温度为45.5℃,相变潜热为28.92 J/g。
(3)TGA分析表明,壁材包覆增加了芯材的热分解温度,降低了分解速率,结果表明,PMMA壁材可有效提高微胶囊的热稳定性和耐热性。
(4)通过热成像得到的数据绘制出调温织物的降温曲线,结果表明经相变微胶囊整理后的调温织物在冷却的过程中可以释放热量。有效降低冷却速度,具有一定的温度调节功能。然而,随着涂层中微胶囊浓度的增加,织物表面上的微胶囊似乎受损并且不均匀。在随后的研究中,可以尝试改善微胶囊的强度并优化涂覆过程以改善这些情况。
参考文献:
[1]王元,刘晓光,杨俊杰,等.相变储能技术的研究进展与应用[J].煤气与热力,2010(9):10-12.
[2]刘光辉.几种相变材料熔化焓的研究[D].北京理工大学,2015.
[3]谷海明.相变储能材料Mg(NO3)2·6H2O的稳定与储热性能研究[D].昆明理工大学,2013.
[4]李刚,孙庆国.无机芯微胶囊相变材料的研究进展[J].无机盐工业,2014,46(10):14-17.
[5]孟令阔.硬脂酸相变微胶囊的制备与表征[D].东华大学,2016.
[6]庞方丽,王瑞,刘星,等.相变微胶囊及其在蓄热调温织物上应用的研究进展[J].天津工业大学学报,2014,33
(3):24-28、33.
[7]陶磊.硫黄微胶囊的制备、表征及应用性能研究[D].青岛科技大学,2016.
[8]辛长征.耐温性相变材料微胶囊的制备及其熔喷纺丝应用研究[D].东华大学,2013.
[9]车迪,陈英.界面聚合法制备新型防蚊微胶囊[J].纺织导报,2018(5):81-85.
[10]梁丰收,陈卫星,马爱洁,等.界面聚合法制备异氰酸酯型微胶囊及其性能[J].高分子材料科学与工程,2018,34(2):150-154.
[11]周宇飞,袁一鸣,仇中柱,等.纳米铝和石墨烯量子点改性的相变微胶囊的制备及特性[J].材料导报,2019,33(6):932-935.
[12]周龙祥,王保明,田玉提,等.二氧化钛包覆石蜡相变微胶囊的制备及表征[J].现代化工,2019,39(3):82-86.
[13]叶泛,黄志明,江夏,等.微悬浮聚合法制备纳米聚甲基丙烯酸甲酯研究[J].长江大学学报(自然科学版),2019,16(1):118-124、10.
[14]信建豪,尚曙玉,王欢欢,等.悬浮聚合法制备同型半胱氨酸分子印迹聚合物微球及应用[J].理化检验(化学分册),2018,54(11):1268-1271.
[15]潘志文,王文利,王秋,等.基于相变微胶囊的功能纺织品制备与应用研究[J].化工新型材料,2018,46(11):256-259.
[16]崔锦峰,张亚斌,张静,等.微胶囊制备技术及其聚合物基功能复合材料研究与应用进展[J].涂料工业,2018,48(11):15-22.
[17]朱振国.具有光致变色功能的相变储能微胶囊的制备及性能[D].天津工业大学,2018.
[18]朱阳倩,汪海平.壁材掺杂纳米TiO2的正十四醇相变微胶囊的制备及性能研究[J].化工新型材料,2019,47(3):134-137、142.
[19]张鑫,党洪洋,龙柱.掺杂钨纳米二氧化钒控温微胶囊的合成与表征[J].塑料工业,2019,47(2):31-36.
[20]Tahan latibris,Mehralime,et al.Fabrication and Perfor⁃mances of Microencapsulated Palmitic Acid with En⁃hanced Thermal Properties[J]Energy&Fuels,2015,1010-1018.
Preparation and Performance Study of LA-SA/MMA Phase Change Microcapsules
LI Xiang,XU Wanping,KE Guizhen
(School of Textile and Engineering,Wuhan Textile University,Wuhan Hubei 430073,China)
Abstract:Phase change microcapsules LA-SA/MMA were prepared by in-situ polymerization method,with methyl methacrylate as the wall material and lauric acid and stearic acid as the core material.Using SEM scanning electron microscopy,differential scanning calorimetry,Malvern particle size analyzer,Fourier transform infrared spectroscopy analyzer,and TG thermogravimetric analyzer,microcapsules with different core to wall ratios were observed and detected.The results showed that microcapsules with acore to wall ratio of 4:5 were spheri-cal in shape,with asmooth and flat surface,but there was slight agglomeration and adhesion phenomenon.The capsule wall has acertain protective effect on the core material,effectively improving the thermal stability of the microcapsule.Microcapsules have excellent durabili-ty and can be reused multiple times.Then,different mass fractions of phase change microcapsules were coated onto SMS fabrics using dry coating technology to produce phase change temperature controlled fabrics.Through thermal imaging,discuss whether the thermal storage and temperature control performance of the fabric treated with phase change microcapsule coating is affected.The results show that the fab-ric after coating treatment has aslower decrease in cooling rate and can cope with changes in external environmental temperature.
Keywords:methyl methacrylate;lauric acid;stearic acid;phase change microcapsules;in-situ polymerization method
(责任编辑:孙婷)
