
SNP遗传力估计方法、影响因素及其在畜禽育种中的应用
2024-09-19 00:00:00段益欣张林云赵永聚
畜牧兽医学报 2024年5期
关键词:影响因素



摘 要: 遗传力是衡量由遗传因素解释的表型方差的比例,在实际遗传力估计中多只考虑加性遗传效应。由于畜禽传统遗传力估计需要的系谱信息完整性和准确性较难以实现,随着基因组学的发展,以全基因组单核苷酸多态性(SNP)进行遗传力估计的方法得到广泛应用。而对SNP遗传力的准确估计有利于了解遗传变异对表型的作用程度,目前已有不少SNP遗传力估计模型被开发利用。本文主要综述了SNP遗传力的概念、常用估计方法及其影响因素,并与传统遗传力估计进行比较,探究了SNP遗传力在畜禽育种中的应用潜力。
关键词: SNP遗传力;遗传力缺失;影响因素;估计模型;畜禽育种
中图分类号:S813
文献标志码:A""" 文章编号:0366-6964(2024)05-1854-12
收稿日期:2023-10-27
基金项目:国家重点研发计划项目(2022YFD1300200);重庆市现代农业产业技术体系(草食牲畜:CQMAITS202313);重庆市科企联合体种质资源收集利用与品种试验项目资助(草食牲畜)资助
作者简介:段益欣(2001-),女,江西于都人,硕士生,主要从事动物遗传育种与繁殖研究,E-mail:duanyx315@163.com
*通信作者:赵永聚,主要从事山羊遗传育种研究,E-mail:zyongju@163.com
The Evaluated Methods and Influencing Factors of SNP Heritability and Its Application
in Farmer Animal Breeding
DUAN" Yixin, ZHANG" Linyun, ZHAO" Yongju*
(Chongqing Engineering Research Center for Herbivores Resource Protection and Utilization,
Chongqing Key Laboratory of Herbivore Science, College of Animal Science and Technology,
Southwest University, Chongqing 400715," China)
Abstract:" Heritability is a measure of the proportion of phenotypic variance explained by genetic factors. In actual heritability evaluation, only additive genetic effects are considered. With the development of genomics, genome-wide single nucleotide polymorphisms (SNPs) have been widely used to evaluate heritability because of the difficulty in achieving the integrity and accuracy of genealogy information that traditional heritability estimation relies on. However, accurate evaluation of SNP heritability is helpful in understanding the extent of genetic variation’s effect on phenotype. Currently, many SNP heritability evaluation models have been developed and utilized. In this paper, the concept, common evaluation methods and influencing factors of SNP heritability are reviewed, and a comparison was made between SNP heritability and the traditional heritability estimation, in order to explore the potential application of SNP heritability in livestock breeding.
Key words: SNP heritability; loss of heritability; influencing factors; estimation model; farmer animal breeding
*Corresponding author:" ZHAO Yongju, E-mail: zyongju@163.com
表型性状通常受到遗传效应和环境效应的共同作用,遗传力是评估遗传与环境效应的关键值,是衡量由遗传因素解释的表型方差的比例[1]。遗传力分为广义遗传力和狭义遗传力,遗传效应包括上位效应、显性效应和加性效应。其中,广义遗传力通常指上位效应、显性效应和加性效应的遗传方差占表型方差的比例,狭义遗传力通常指加性遗传效应解释的表型方差的比例[1-3]。传统遗传力的估计通常针对狭义遗传力进行,主要基于家庭或系谱数据,利用混合线性模型(MLM)进行方差组分的估计,从而进行加性遗传效应的评估。作为遗传效应影响表型的重要参数之一,遗传力常被用作遗传连锁和遗传关联研究的基础[4],其应用主要包括了解遗传能力、确定疾病遗传风险、比较遗传起源及遗传相关,甚至可以辅助育种值准确估计从而促进基因组选择等[1,3,5]。较高遗传力表明遗传因素在影响性状表型或疾病上发挥较高作用[6],因此,在畜禽育种中遗传力的准确估计具有重要意义,性状的遗传力高低将影响其选种、选育的方式方法,而遗传力的准确估计也是性状遗传评估的基础[7]。
由于传统的遗传力估计是基于系谱信息来构建亲缘关系矩阵(A阵),并结合MLM进行的,这对系谱信息的准确性以及完整性都有较高的要求[8]。而目前,大多数养殖场存在系谱信息记录不完善和错误等情况,致使传统的遗传力估计方法存在一定的局限性。随着测序技术的快速发展,数以百万计的SNPs得以有效挖掘,利用SNP进行遗传力估计成为了一种流行趋势。Yang等[9]开发了一种基于全基因组SNP进行的估计加性遗传变异比例方法,该方法利用全基因组SNP分子标记构建相关性矩阵(GRM),从而进行遗传力的估计。本文主要对SNP遗传力估计的概念、估计方法及其影响因素进行综述,并与传统遗传力估计进行比较以了解SNP遗传力估计的重要意义,探究SNP遗传力在畜禽育种中的应用潜力。
1 SNP遗传力概述
1.1 SNP遗传力概念
利用SNP分子标记进行加性遗传方差的估计以解释表型变异的比例即为SNP遗传力(h2SNP)。SNP遗传力通常利用REML法拟合方差分量模型进行估计[10]。一般SNP遗传力估计分为两类:基于全基因组关联分析(GWAS)结果和基于全基因组SNP[5]。前者是利用GWAS研究中显著性SNP进行遗传力估计,后者是利用了全基因组所有的SNP。全基因组方法表明了大部分遗传变异是加性遗传变异,且其中的约1/3可由SNP捕获[11],常见的SNP可以解释大部分性状的遗传性[12]。
随着全基因组测序技术和GWAS的发展,目前已经能够准确测定数以百万计的SNP的基因型以及挖掘复杂性状相关的分子标记及基因。但是研究发现,基于GWAS结果只能解释一小部分的遗传力,而有很大一部分的遗传力无法解释,这一部分的遗传力称之为“消失遗传力”或“遗传力缺失”[13]。消失遗传力的划分主要为隐藏的遗传力和依旧消失的遗传力[14],隐藏的遗传力通常指全基因组SNP遗传力与GWAS遗传力之间的差异,主要因小效应的标记无法检测所致,可通过增加样本量的方式来降低;依旧消失的遗传力通常指利用GWAS也无法捕获的变异,例如一些罕见的变异,可以通过增加变异来源、估计多种遗传方差来降低。
对于“消失遗传力”的解释主要包括:变异与SNPs不完全连锁不平衡(LD)导致遗传力估计偏低;具有较大效应的罕见变异解释了表型方差;影响较小的常见变异(MAFgt;1%)无法在全基因组显著性水平上检测到;环境影响使得基于系谱研究的遗传力估计值偏高;遗传相互作用使总遗传力估计偏高[9,11,13,15-16]。大多数基因水平的遗传力由常见变异主导,但有研究从罕见或低频变异(0.5%lt;MAFlt;1%)中确定了影响某些性状的基因,其遗传力不为零[17]。Zhou等[18]在番茄图形泛基因组的构建研究中估计了不同变异对遗传力的贡献,发现SNP对遗传力的贡献值并非最高,而由于DNA片段发生插入、缺失、重复等遗传变异所引起的基因组结构变异(SV)对于遗传力也有较大的贡献,其作用不可忽视,这可能也是影响缺失遗传力的一大因素,即表明结构变异可以捕获一部分缺失的遗传力。另有研究指出,尽管大多数变异具有累加效应,但非加性遗传效应对遗传力估计的影响仍不可忽视[19]。Alipanah等[20]对苏格兰绵羊进行体重性状的遗传参数估计研究发现,加性和非加性效应的联合模型对遗传参数的估计研究更为准确。此外,Chen等[21]对人双胞胎个体的研究也指出了显性效应在遗传估计中的作用可能被大大低估。尽管有研究发现泛基因组开放阅读框可以为缺失遗传力的研究进行潜在补充[22],但目前遗传力缺失的具体机制与原因仍未得到完全的解释,而由于“缺失遗传力”存在着多样性,大部分研究者着重于对基因组和遗传因素控制复杂表型的机制进行研究。
基于系谱信息的遗传力估计受到基因-环境互作效应的影响,其估计值可能偏高[15],而GWAS研究中发现的与性状相关的SNPs只能解释一小部分遗传力,其遗传力估计值偏低。因此,利用全基因组SNP进行遗传力的准确估计对于遗传评估、遗传改良、缺失遗传力的研究以及GWAS研究[23]皆至关重要。
1.2 传统遗传力估计与SNP遗传力估计的比较
相较于传统遗传力估计方法,系谱信息难以保证其准确性和完整性,而基于全基因组标记的SNP遗传力估计恰恰可以避免这一问题。为进一步提高遗传力估计的准确性和完整性,基于SNP标记进行的遗传力估计研究日益增多,不少研究者对传统遗传力估计法与SNP遗传力估计法进行了比较研究[13,24-27]。有研究指出,与系谱信息相比,由基因组信息估计的人类复杂性状的遗传力估计值偏低[13],即所谓的“遗传力缺失”。Khanzadeh等[28]基于系谱信息和基因组信息对奶牛产奶性状进行遗传力估计的研究指出,基于谱系的遗传力估计值要高于基因组遗传力估计值。然而仍有不少研究结果与其存在差异。
有研究指出,相较于基于系谱数据的遗传力估计值,基于单核苷酸多态性的遗传力估计能够捕获更大的加性遗传方差比例[29],且具有较高的准确性和稳定性[30]。常瑶等[24]基于系谱信息以及基因组信息对不同于月龄的荷斯坦牛后备牛的体重性状进行了遗传参数估计,研究指出,利用系谱-基因组信息估计的遗传参数更准确、稳定。Perrier等[30]基于系谱信息、全基因组信息、基因组矫正的系谱信息,对小样本的蓝山雀进行4个性状的遗传参数估计研究,结果发现与基于社会系谱信息(PED)估计的遗传力相比,使用遗传关系矩阵(GRM)估计的遗传力估计值略高,利用基因组信息进行的校正系谱信息(PED校正)估计的遗传力处于中间值,表明由于不正确的系谱信息以及系谱信息含量低于遗传关系矩阵(GRM),致使PED遗传力略有被低估,而遗传关系矩阵(GRM)具有更高的精确度和更高的信息量。另有研究指出,虽然基因组遗传力估计具有相对较大的标准误差,但系谱信息和基于SNP标记估计的遗传力差异不大[25]。Guo等[26]对扇贝的四个生长性状进行遗传力估计,比较了GCTA法(基于基因组SNP)和传统选择性育种法(选择反应与选择差的比值)估计遗传力的差异,结果发现两者的估计值差异不大。Ogawa等[31]对日本黑牛胴体性状进行研究发现,基于基因组信息的遗传力估计值与传统系谱法估计值相似,即表明大部分加性遗传变异可以由SNP解释。Aase等[32]研究也指出,基于基因组信息进行的定量遗传参数估计与系谱估计值相似。Bérénos等[27]通过比较系谱和全基因组两种方法估计的遗传力也发现其遗传力估计值相似,但基因组法所估计的遗传力其标准差较低,且包含了更多的信息。
对于基于系谱数据和基于SNP标记进行的遗传力估计比较研究似乎并没有统一确定的结果,这可能与系谱准确性[30]、SNP密度[26,33]、估计算法(模型)[34]的差异有关。但毫无疑问的是,相较于系谱信息的较难收集及其完整性、准确性不明,基于基因组SNP标记进行的遗传力估计研究的准确性、精确性及稳定性更佳。而由于考虑的遗传变异有限以及标记间的连锁不平衡,SNP遗传力估计结果与实际遗传力间仍存在一定的差异。
另外,遗传参数的估计需要大的样本量和有关个体间完整、准确的系谱信息。对于那些只能在较小样本中表型化或具有成本效益的数量性状,如罕见病、行为性状等,可能较难获得足够大的样本量以满足估计遗传参数的要求,使用SNP进行遗传力估计比系谱法的遗传力估计效果更佳[29]。
2 SNP遗传力估计方法
通常利用线性回归模型假设表型向量(y)与基因型矩阵(X)间的关系:
y=Xβ+ε
(1)
上式(1)中,y表示表型向量,X表示n×p的基因型矩阵(n为样本数量,p为SNP数量),β表示遗传效应大小,ε表示残差向量。基因型利用0、1、2进行编码,表示参考等位基因的数量。当ngt;p时,遗传力由Var(Xβ)/Var(y)估计,其中Var表示样本方差;当nlt;p时,上式(1)成为了一个不确定的系统,因此需要对SNP效应大小进行模型假设[1-2]。
目前已有不少可用于SNP遗传力估计的关于效应大小的模型假设,常用的一些模型假设如下:
基因组相关性矩阵限制最大似然法(GREML)。混合线性模型(MLM)最初由Yang等[9]应用于SNP遗传力估计。其假设所有SNP具有非零效应,且效应(β)大小遵循正态分布:
βj~N(0,σ2β)(2)
y=Xf+Zα+e
(3)
上式(3)中,y表示性状的表型,f表示固定效应(非遗传),α表示随机加性效应,e表示残差效应,X、Z分别为对应的结构矩阵。基于混合线性模型进行的SNP遗传力估计方法可以分为两类[35]:一类是将SNP效应作为固定效应加入到线性模型中[36],另一类是将SNP效应作为随机效应[9]。固定效应模型假设每个SNP在种群内的不同样本中的影响是一致的,而在随机效应模型中,SNP的影响可能在不同样本中发生变化[36]。
SNP遗传力估计值为:h2SNP=σ2gσ2g+σ2e,其中加性遗传方差定义为:σ2g=p×σ2β。基于全基因组数据,应用MLM模型可能会导致低估了具有常见的潜在因果变异的性状的SNP遗传力[1]。因此,总加性遗传效应的方差分量和残差效应的方差分量可以利用GCTA软件[37]基于约束最大似然法(REML)进行估算,即GREML法。GREML方法在估计时考虑了LD,并且其应用于不相关的个体样本,因此SNP遗传力不太可能被常见的环境影响所混淆[38]。
贝叶斯变量选择回归法(BVSR)。BVSR法假设在所有遗传变异中影响表型的变异相对较少[39]。假设每个SNP效应大小服从点-正态分布:
βj~πN(0,σ2β)+(1-π)δ0
(4)
上式(4)中,σ2β表示正态分布中的方差分量,π表示非零SNP效应比例,而当比例为1-π时,SNP效应为零,δ0为零点的质点。SNP遗传力估计为:h2SNP=πσ2βπσ2β+σ2e,利用马尔可夫链蒙特卡罗(MCMC)算法获得参数样本。不少研究利用REML进行遗传力估计,利用贝叶斯模型进行SNP遗传力估计相对较少,但有研究对比了REML和贝叶斯法(Bayes)来估计SNP遗传力,指出利用贝叶斯模型进行高遗传力性状的SNP遗传力估计也同样具有较高的准确性,但对于较低遗传力的性状,所有模型都难以估计真实的遗传力[40]。
当小部分SNP对性状具有非零效应时,BVSR在估计SNP遗传力方面更准确;相反,当大部分SNP对性状具有效应时,利用MLM估计SNP遗传力更为准确。
贝叶斯系数线性混合模型(BSLMM)。通常由于表型性状的遗传结构是未知的,所以对于模型的选择也无法确定。因此,Zhou等[41]将LMM和BVSR两种模型进行综合,提出了贝叶斯稀疏线性混合模型(BSLMM),假设效应服从两个正态分布的混合:
βj~πN(0,σ2a+σ2b)+(1-π)N(0,σ2b)
(5)
假设所有SNP至少有一个小的影响其方差为σ2b,当比例为π 时,方差分量为σ2a,当σ2b为0时,相当于BVSR模型;当π为0时,相当于LMM模型。SNP遗传力估计近似于pπσ2a+σ2bpπσ2a+σ2b+σ2e,依靠大都会-黑斯廷斯(MH)算法执行后验。
连锁不平衡调整亲属关系(LDAK)。由于LD的存在,BVSR,GREML和BSLMM法可能会对高LD的变异的遗传力估计值高估,而低估了低LD区域的遗传力估计。Speed等[42]提出了一种新方法LDAK,它考虑了LD,假设由罕见变异解释的方差比由常见变异解释的方差要大。假设效应大小遵循以下等式:
βj~N(0,σ2j),σ2j∝(fj(1-fj))1+α×ωj×rj
(6)
上式(6)中,fj是指SNPj的次等位基因频率;ωj是SNP权重,是SNPj的连锁不平衡分数倒数的函数。LDAK法利用LD调整GRM,即通过LD加权模型,减少了潜在偏差,提高了遗传力估计的精确性[43]。但LDAK中的LD加权增加了稀有变异的比重,从而导致对罕见变异遗传力的高估[6]。
连锁不平衡分数回归(LDSC)。根据GWAS结果汇总统计显著性SNP数据估计SNP遗传力的常用方法是LDSC[44]。
E(X2|li)=nlih2SNPp+na+1
(7)
上式(7)中,n是样本数量,p是 SNP 的数量,a是衡量混杂偏差的贡献,给定SNP的X2关联统计值反映LD中所有SNP与该SNP的关联,li是SNPi的LD得分,即LDSC通过回归 GWAS 测试统计数据X2每个 SNP LD 分数li估计h2SNP。
除了以上被列举出来的方法以外,还有不少其它的估计模型,例如应用于数量性状的广义随机效应模型(GRE)[45]、相关性不平衡回归(RDR)[46]、动态贝叶斯高斯过程模型(dynBGP)[47],应用于病例对照研究和计数表型的责任阈值模型-REML[48]、PCGC回归[49]、PQLseq[50]以及应用于汇总统计的MQS法[51]、GWAS遗传力估计器(GWASH)[36]等。目前,基于SNP的遗传力研究中,GCTA法(也称GREML法)被较为广泛的应用于数量性状研究中,而连锁不平衡回归分析法(LDSC)被最为广泛的应用于汇总统计数据当中[52]。BVSR和BSLMM法除了可以估计SNP遗传力外,也可用于基因组预测研究中。表1为 h2SNP估计方法总括表。
3 SNP遗传力估计的影响因素
遗传力不是表型性状的固有属性,其取决于测定表型的样本群体[17,52],它会随着群体差异(包括年龄、性别、季节等)、环境差异而变化。遗传参数的估计对于样本量的要求也较高,通常通过增加样本量的方式减少抽样方差[54]。而基于不同平台检测的SNP位点数量不同[30],因果变异的连锁不平衡、次等位基因频率[9,55-56],群体分层[57]以及估计的模型不同[34],会导致SNP遗传力估计值间可能存在差异。
SNP遗传力估计研究依赖于基因组信息,SNP标记密度的增加可以提高遗传力估计的精确度。Bérénos等[33]对野生绵羊种群5个成年体型性状进行遗传结构分析发现,其基因组遗传力范围从前腿0.26到下颌0.53,且不同体型性状具有不同遗传结构,而密集的SNP有助于了解野生种群基因座数量特征的选择。Guo等[26]的研究也发现SNP密度会影响遗传力估计值,当标记密度足够高时,利用GCTA法可进行准确的遗传力估计。但也有研究发现SNP密度的增加对遗传力估计的影响效果并不显著[40,58],James等[59]利用两种不同SNP密度对3个年龄阶段(初生、羔羊、成年)的野生绵羊的5个性状(体重、前腿长、后腿长、掌骨长度、下颌长度)进行SNP遗传力估计,并进行GWAS分析,发现不同SNP密度并不会影响遗传力的估计,但在高密度SNP数据发现了一些新的SNP-性状关联。另外,倪萍等[8]对仿刺参的疣足数量进行SNP遗传力估计时发现50K SNP密度下的遗传力估计已达稳定。这表明不同物种不同性状的SNP遗传力估计对SNP密度的要求不一致,不同的SNP密度对遗传力估计存在着一定影响,而当标记密度达到一定水平时,这种影响将达到稳定,而大多数遗传方差都可以通过高密度的SNP捕获[55]。
SNP间的LD会致使SNP遗传力估计偏倚,因此进行SNP遗传力估计时一般建议考虑LD,目前有不少研究将LD加权进入模型中以保证SNP遗传力估计的准确性[29,42,60]。Ren等[61]利用LD调整亲缘关系(LDAK)和LD分层多组分(LDS)模型控制区域间LD异质性,并与没有LD控制的经典模型进行比较,发现GCTA-LDS模型和LDAK-LDS模型都能有效消除区域间LD异质性的不利影响,从而提高遗传力估计的无偏性和基因组预测的准确性;另外,研究发现对于高密度SNP数据,区域间LD异质性的不利影响更为明显。次等位基因频率(MAF)与LD相互依赖,高MAF的数量性状位点(QTL)间的LD程度高于低MAF的QTLs间,通常因果变异的LD会致使遗传力估计偏差,而MAF可作为其不完全指标[42]。另外,当根据MAF进行SNP标记筛选质控时,考虑的SNP标记位点会有所不同,从而会引起遗传力估计的差异性。
SNP遗传力估计会受到许多因素的影响,在应用过程中往往需要根据实际情况考虑不同的方法进行相关的研究估计。尽管目前的模型方法估计SNP遗传力存在一定的偏差,但整体而言,大多数方法模型都是可靠稳健的[45]。
4 SNP遗传力估计在畜禽育种中的应用
遗传力是反映性状遗传能力大小的重要遗传参数,遗传力估计的准确度对畜禽育种工作具有重要的意义,通常可以根据遗传力的大小选择适合的育种方法,为畜禽育种增效。传统遗传力估计依赖于亲缘关系较近的个体间系谱信息的完整性及准确性,并且由于受到环境因素的作用,其估计值往往偏高。随着SNP标记估计遗传力方法的改善,可以对许多复杂性状进行较为准确的SNP遗传力估计,复杂性状的遗传学研究也得到了显著进展。
在畜禽育种方案设计中,改良性状的遗传力和加性遗传效应对获得较佳的育种选择至关重要[62],因此,基于基因组信息进行准确的遗传参数估计十分重要。阮栋林等[63]对杜洛克种猪生长和体尺性状进行遗传参数估计研究时,利用了SNP芯片数据进行估计研究,并加入了环境因素和群体间混杂因素的影响,结果指出该群体的生长性状属于中高等遗传力性状,在选育过程中较易、较快获得遗传进展,而大部分体尺性状属于低遗传力性状,较易受到环境因素的影响,说明在选育过程中需结合考虑不同的选择方法,进而加快遗传改良进展,也为杜洛克猪的选育提供了一定的理论基础。羊毛性状作为绵羊重要经济性状之一,是绵羊选育过程中考虑的一个重要指标。Arzik等[64]基于基因组信息对阿卡拉曼羊的5个羊毛性状进行遗传参数估计,发现6个羊毛性状的遗传力范围包括中等到高等,结合GWAS研究挖掘出与羊毛性状显著相关的1个基因和6个SNPs,为提高该品种羊毛质量和产量的育种计划提供帮助。另外,通过考虑SNP密度和性状遗传力来设计基因组选择策略,在育种过程中实现可持续的遗传增益和控制近交水平十分重要[65]。Zhang等[66]通过研究肉脂型与瘦肉型两种群体的北京鸭肉质性状发现,肉脂型与瘦肉型北京鸭在整体基因组水平具有遗传可区分性,其肉质性状的遗传力也存在差异性,即两个种群间不同选择方向导致了更高水平的遗传分化,结合基因组选择,可为鸭育种改良提供帮助。Rostamzadeh等[67]对日本黑牛胴体性状进行基因组遗传力及相关性分析指出,其胴体性状的遗传力为中等至高等范围,胴体性状之间存在相当大的遗传变异和较好的基因组相关性,即通过基因组选择可以同时获得性状的遗传改良,研究还发现样本量以及个体之间的相关性不同,可能会导致基因组遗传力和胴体性状之间相关性估计值的不同。
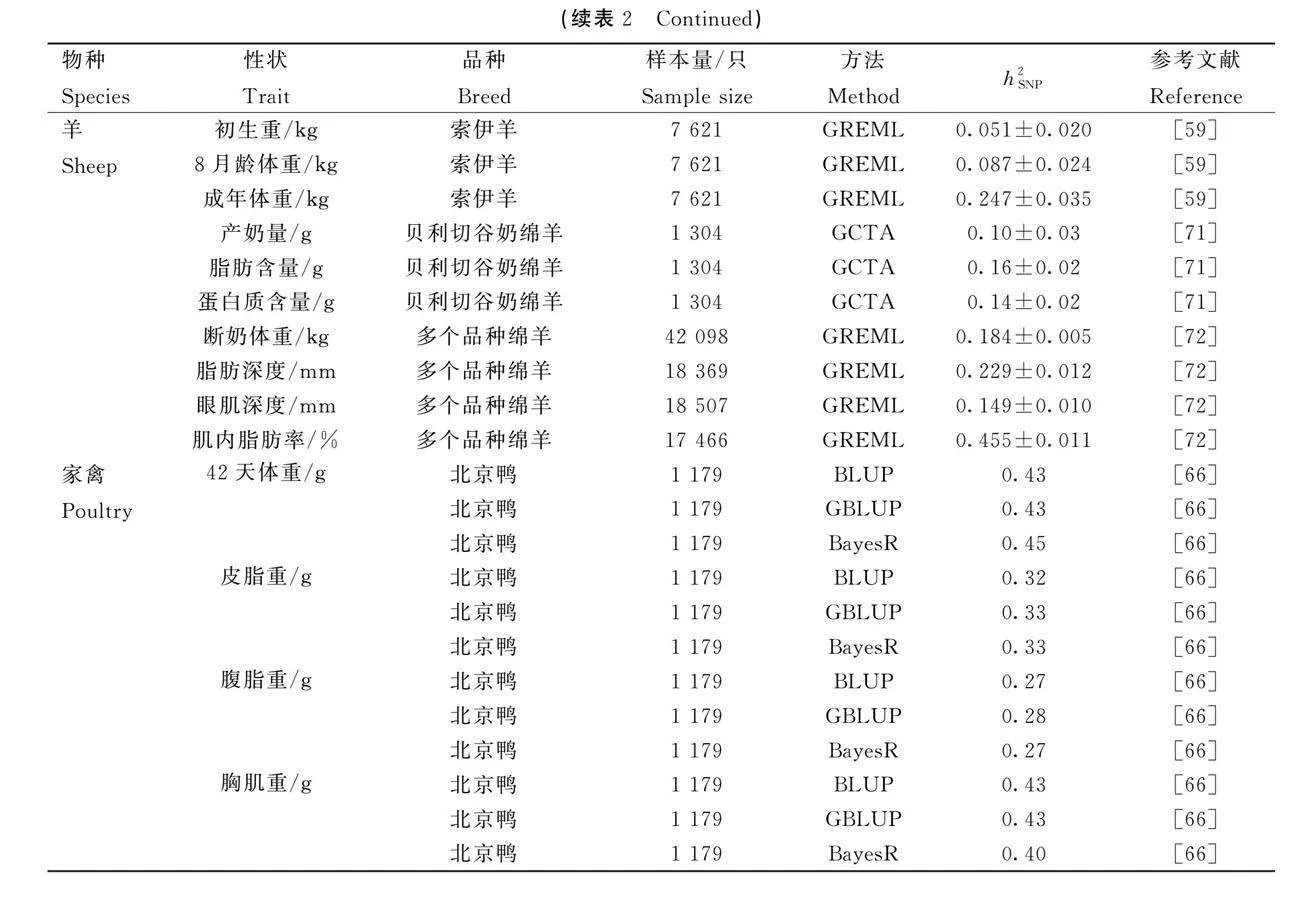
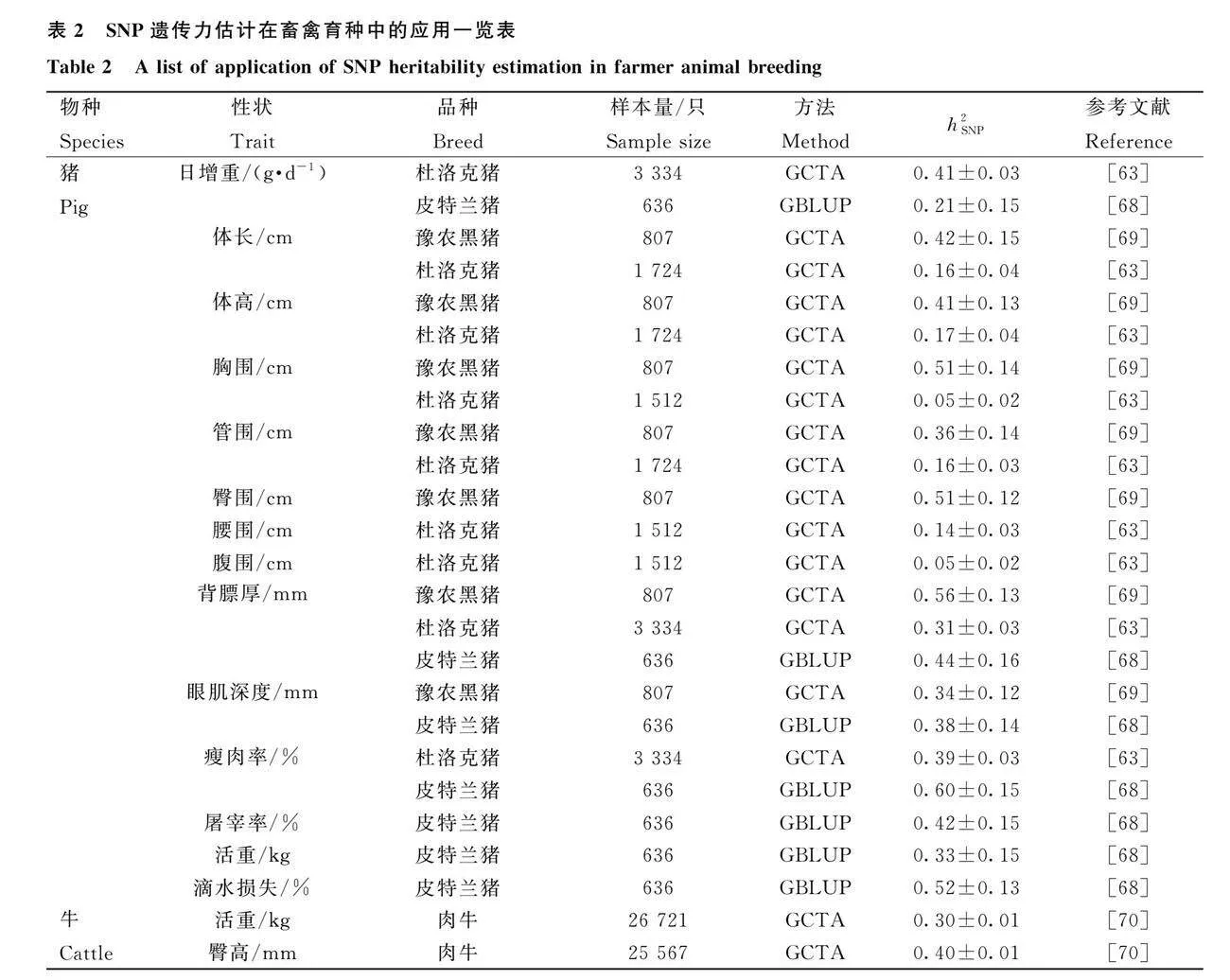
通常SNP遗传力并不会单独进行研究估计,往往会结合GWAS分析、基因组育种值估计并对基因组进行选择,共同为动物的育种工作提供一定的理论依据。表2汇总了目前不同畜禽重要性状的SNP遗传力估计的研究情况。
5 总 结
SNP遗传力估计能克服传统遗传力估计受系谱信息完整性的限制难题,为遗传参数准确估计提供了可能,并结合当前如火如荼的基因组研究,为畜禽育种工作提供更有效的帮助。值得注意的是,目前基于SNP标记进行的遗传力估计结果与实际遗传力间仍存在一定差异,其准确性也有待更进一步的提高。现有的SNP遗传力估计模型所考虑的因素并不充分,其估计值仍存在一定的偏差,且仍需有足够大的样本,因此SNP遗传力估计的实际应用受到一定的限制。其次,大部分的复杂性状存在着隐藏遗传力,而基于SNP分子标记的遗传力估计尚存在一定的局限性,并不能完全考虑所有的变异,因此在遗传力估计中应更多的考虑各种变异的存在。再者,目前的SNP遗传力估计模型大多只考虑了加性遗传效应,而忽略了显性效应和互作效应。因此,随着测序技术、方法以及SNP遗传力估计模型的不断发展与成熟,有望建立更为准确的估计模型,使基于全基因组信息进行SNP遗传力无偏估计得到更广泛应用,并尽可能充分考虑各种遗传、环境等因素的影响,为畜禽育种提供有效帮助。
参考文献(References):
[1] ZHU H H,ZHOU X.Statistical methods for SNP heritability estimation and partition:a review[J].Comput Struct Biotechnol J,2020,18:1557-1568.
[2] TANG M S,WANG T,ZHANG X F.A review of SNP heritability estimation methods[J].Brief Bioinform,2022,23(3):bbac067.
[3] VISSCHER P M,HILL W G,WRAY N R.Heritability in the genomics era-concepts and misconceptions[J].Nat Rev Genet,2008,9(4):255-266.
[4] FRIEDMAN N P,BANICH M T,KELLER M C.Twin studies to GWAS:there and back again[J].Trends Cogn Sci,2021,25(10): 855-869.
[5] MAYHEW A J,MEYRE D.Assessing the heritability of complex traits in humans:methodological challenges and opportunities[J]. Curr Genomics,2017,18(4):332-340.
[6] SRIVASTAVA A K,WILLIAMS S M,ZHANG G.Heritability estimation approaches utilizing genome-wide data[J].Curr Protoc,2023,3(4):e734.
[7] PIZARRO INOSTROZA M G,LANDI V,NAVAS GONZLEZ F J,et al.Does the acknowledgement of αS1-casein genotype affect the estimation of genetic parameters and prediction of breeding values for milk yield and composition quality-related traits in Murciano-Granadina?[J].Animals (Basel),2019,9(9):679.
[8] 倪 萍,任 强,王 静,等.仿刺参疣足数量SNP遗传力评估[J].渔业科学进展,2021,42(3):68-76.
NI P,REN Q,WANG J,et al.Estimating SNP heritability for papillae number in sea cucumber[J].Progress in Fishery Sciences,2021,42(3):68-76.(in Chinese)
[9] YANG J,BENYAMIN B,MCEVOY B P,et al.Common SNPs explain a large proportion of the heritability for human height[J]. Nat Genet, 2010,42(7):565-569.
[10] LIN Z T,SEAL S,BASU S.Estimating SNP heritability in presence of population substructure in biobank-scale datasets[J]. Genetics,2022,220(4):iyac015.
[11] UZZAMAN M R,PARK J E,LEE K T,et al.Whole-genome association and genome partitioning revealed variants and explained heritability for total number of teats in a Yorkshire pig population[J].Asian-Australas J Anim Sci,2018,31(4):473-479.
[12] SHIN D,PARK K D,KA S,et al.Heritability estimated using 50K SNPs indicates missing heritability problem in Holstein breeding[J].Genomics Inform,2015,13(4):146-151.
[13] MANOLIO T A,COLLINS F S,COX N J,et al.Finding the missing heritability of complex diseases[J].Nature,2009, 461(7265):747-753.
[14] WITTE J S,VISSCHER P M,WRAY N R.The contribution of genetic variants to disease depends on the ruler[J].Nat Rev Genet,2014,15(11):765-776.
[15] YANG J,BAKSHI A,ZHU Z H,et al.Genetic variance estimation with imputed variants finds negligible missing heritability for human height and body mass index[J].Nat Genet,2015,47(10):1114-1120.
[16] TENESA A,HALEY C S.The heritability of human disease:estimation,uses and abuses[J].Nat Rev Genet,2013,14(2):139-149.
[17] BURCH K S,HOU K C,DING Y,et al.Partitioning gene-level contributions to complex-trait heritability by allele frequency identifies disease-relevant genes[J].Am J Hum Genet,2022,109(4):692-709.
[18] ZHOU Y,ZHANG Z Y,BAO Z G,et al.Graph pangenome captures missing heritability and empowers tomato breeding[J].Nature,2022,606(7914):527-534.
[19] LPEZ-CORTEGANO E,CABALLERO A.Inferring the nature of missing heritability in human traits using data from the GWAS catalog[J].Genetics,2019,212(3):891-904.
[20] ALIPANAH M,ROUDBARI Z,MOMEN M,et al.Impact of inclusion non-additive effects on genome-wide association and variance’s components in Scottish black sheep[J].Anim Biotechnol,2023,34(8):3765-3773.
[21] CHEN X,KUJA-HALKOLA R,RAHMAN I,et al.Dominant genetic variation and missing heritability for human complex traits:insights from twin versus genome-wide common SNP models[J].Am J Hum Genet,2015,97(5):708-714.
[22] LI Z C,SIMIANER H.Pan-genomic open reading frames:a potential supplement of single nucleotide polymorphisms in estimation of heritability and genomic prediction[J].PLoS Genet,2020,16(8):e1008995.
[23] SPEED D,O’BRIEN T J,PALOTIE A,et al.Describing the genetic architecture of epilepsy through heritability analysis[J].Brain,2014,137(10):2680-2689.
[24] 常 瑶,苏国生,李艳华,等.基于系谱和基因组信息估计荷斯坦青年母牛体重性状遗传参数[J].畜牧兽医学报,2022,53(11):3759-3768.
CHANG Y,SU G S,LI Y H,et al.Estimating genetic parameters for body weights using pedigree and genotype-pedigree based approaches in Holstein heifers[J].Acta Veterinaria et Zootechnica Sinica,2022,53(11):3759-3768.(in Chinese)
[25] JOERGENSEN D,MADSEN P,ROPSTAD E O,et al.Heritability estimates of distichiasis in Staffordshire bull terriers using pedigrees and genome-wide SNP data[J].Acta Vet Scand,2022,64(1):30.
[26] GUO H B,ZENG Q F,LI Y P,et al.Estimating realized heritability for growth in Zhikong scallop (Chlamys farreri) using genome-wide complex trait analysis[J].Aquaculture,2018,497:103-108.
[27] BRNOS C,ELLIS P A,PILKINGTON J G,et al.Estimating quantitative genetic parameters in wild populations:a comparison of pedigree and genomic approaches[J].Mol Ecol,2014,23(14):3434-3451.
[28] KHANZADEH H,GHAVI HOSSEIN-ZADEH N,GHOVVATI S.A meta-analysis of the gap between pedigree-based and genomic heritability estimates for production traits in dairy cows[J].Livest Sci,2022,263:105000.
[29] ZIMMERMANN E,DISTL O.SNP-based heritability of osteochondrosis dissecans in hanoverian warmblood horses[J].Animals (Basel),2023,13(9):1462.
[30] PERRIER C,DELAHAIE B,CHARMANTIER A.Heritability estimates from genomewide relatedness matrices in wild populations:application to a passerine,using a small sample size[J].Mol Ecol Resour,2018,18(4):838-853.
[31] OGAWA S,MATSUDA H,TANIGUCHI Y,et al.Estimation of the autosomal contribution to total additive genetic variability of carcass traits in Japanese Black cattle[J].Anim Sci J,2022,93(1):e13710.
[32] AASE K,JENSEN H,MUFF S.Genomic estimation of quantitative genetic parameters in wild admixed populations[J].Methods Ecol Evol,2022,13(5):1014-1026.
[33] BRNOS C,ELLIS P A,PILKINGTON J G,et al.Heterogeneity of genetic architecture of body size traits in a free-living population[J].Mol Ecol,2015,24(8):1810-1830.
[34] SPEED D,KAPHLE A,BALDING D J.SNP-based heritability and selection analyses:improved models and new results[J]. Bioessays,2022,44(5):2100170.
[35] MIN A L,THOMPSON E,BASU S.Comparing heritability estimators under alternative structures of linkage disequilibrium[J]. G3 (Bethesda),2022,12(8):jkac134.
[36] SCHWARTZMAN A,SCHORK A J,ZABLOCKI R,et al.A simple,consistent estimator of SNP heritability from genome-wide association studies[J].Ann Appl Stat,2019,13(4):2509-2538.
[37] YANG J,LEE S H,GODDARD M E,et al.GCTA:a tool for genome-wide complex trait analysis[J].Am J Hum Genet,2011,88(1): 76-82.
[38] YANG J,ZENG J,GODDARD M E,et al.Concepts,estimation and interpretation of SNP-based heritability[J].Nat Genet,2017,49(9):1304-1310.
[39] GUAN Y T,STEPHENS M.Bayesian variable selection regression for genome-wide association studies and other large-scale problems[J].Ann Appl Stat,2011,5(3):1780-1815.
[40] KRAG K,JANSS L L,SHARIATI M M,et al.SNP-based heritability estimation using a Bayesian approach[J]. Animal,2013,7(4):531-539.
[41] ZHOU X,CARBONETTO P,STEPHENS M.Polygenic modeling with Bayesian sparse linear mixed models[J].PLoS Genet, 2013, 9(2):e1003264.
[42] SPEED D,HEMANI G,JOHNSON M R,et al.Improved heritability estimation from genome-wide SNPs[J].Am J Hum Genet, 2012, 91(6):1011-1021.
[43] SPEED D,CAI N,The UCLEB Consortium,et al.Reevaluation of SNP heritability in complex human traits[J].Nat Genet,2017, 49(7):986-992.
[44] BULIK-SULLIVAN B K,LOH P R,FINUCANE H K,et al.LD Score regression distinguishes confounding from polygenicity in genome-wide association studies[J].Nat Genet,2015,47(3):291-295.
[45] HOU K C,BURCH K S,MAJUMDAR A,et al.Accurate estimation of SNP-heritability from biobank-scale data irrespective of genetic architecture[J].Nat Genet,2019,51(8):1244-1251.
[46] YOUNG A I,FRIGGE M L,GUDBJARTSSON D F,et al.Relatedness disequilibrium regression estimates heritability without environmental bias[J].Nat Genet,2018,50(9):1304-1310.
[47] ARJAS A,HAUPTMANN H,SILLANPM J.Estimation of dynamic SNP-heritability with Bayesian Gaussian process models[J].Bioinformatics,2020,36(12):3795-3802.
[48] LEE S H,WRAY N R,GODDARD M E,et al.Estimating missing heritability for disease from genome-wide association studies[J].Am J Hum Genet,2011,88(3):294-305.
[49] GOLAN D,LANDER E S,ROSSET S.Measuring missing heritability:inferring the contribution of common variants[J].Proc Natl Acad Sci U S A,2014,111(49):E5272-E5281.
[50] SUN S Q,ZHU J Q,MOZAFFARI S,et al.Heritability estimation and differential analysis of count data with generalized linear mixed models in genomic sequencing studies[J].Bioinformatics,2019,35(3):487-496.
[51] ZHOU X.A unified framework for variance component estimation with summary statistics in genome-wide association studies[J].Ann Appl Stat,2017,11(4):2027-2051.
[52] GE T,CHEN C Y,NEALE B M,et al.Phenome-wide heritability analysis of the UK Biobank[J].PLoS Genet,2017,13(4): e1006711.
[53] PALLA L,DUDBRIDGE F.A fast method that uses polygenic scores to estimate the variance explained by genome-wide marker panels and the proportion of variants affecting a trait[J].Am J Hum Genet,2015,97(2):250-259.
[54] VISSCHER P M,GODDARD M E.A general unified framework to assess the sampling variance of heritability estimates using pedigree or marker-based relationships[J].Genetics,2015,199(1):223-232.
[55] UEMOTO Y,SASAKI S,KOJIMA T,et al.Impact of QTL minor allele frequency on genomic evaluation using real genotype data and simulated phenotypes in Japanese Black cattle[J].BMC Genet,2015,16:134.
[56] EVANS L M,TAHMASBI R,VRIEZE S I,et al.Comparison of methods that use whole genome data to estimate the heritability and genetic architecture of complex traits[J].Nat Genet,2018,50(5):737-745.
[57] DANDINE-ROULLAND C,BELLENGUEZ C,DEBETTE S,et al.Accuracy of heritability estimations in presence of hidden population stratification[J].Sci Rep,2016,6:26471.
[58] GODDARD M E,HAYES B J.Mapping genes for complex traits in domestic animals and their use in breeding programmes[J].Nat Rev Genet,2009,10(6):381-391.
[59] JAMES C,PEMBERTON J M,NAVARRO P,et al.The impact of SNP density on quantitative genetic analyses of body size traits in a wild population of Soay sheep[J].Ecol Evol,2022,12(12):e9639.
[60] MATHEW B,LON J,SILLANP M J.A novel linkage-disequilibrium corrected genomic relationship matrix for SNP-heritability estimation and genomic prediction[J].Heredity (Edinb),2018,120(4):356-368.
[61] REN D Y,CAI X D,LIN Q,et al.Impact of linkage disequilibrium heterogeneity along the genome on genomic prediction and heritability estimation[J].Genet Sel Evol,2022,54(1):47.
[62] GUL S,ARZIK Y,KIZILASLAN M,et al.Heritability and environmental influence on pre-weaning traits in Kilis goats[J].Trop Anim Health Prod,2023,55(2):85.
[63] 阮栋林,陈 悦,庄站伟,等.基于SNP芯片的杜洛克种猪生长和体尺性状遗传参数估计及相关分析[J].家畜生态学报,2023,44(2):19-24.
RUAN D L,CHEN Y,ZHUANG Z W,et al.Genetic parameter estimation and correlation analysis of growth traits and body traits in Duroc pigs based on SNP chip[J].Journal of Domestic Animal Ecology,2023,44(2):19-24.(in Chinese)
[64] ARZIK Y,KIZILASLAN M,BEHREM S,et al.Genome-wide scan of wool production traits in akkaraman sheep[J].Genes (Basel),2023,14(3):713.
[65] ZHENG X,ZHANG T L,WANG T Z,et al.Long-term impact of genomic selection on genetic gain using different SNP density[J].Agriculture,2022,12(9):1463.
[66] ZHANG F,ZHU F,YANG F X,et al.Genomic selection for meat quality traits in Pekin duck[J].Anim Genet,2022,53(1):94-100.
[67] ROSTAMZADEH MAHDABI E,TIAN R G,LI Y,et al.Genomic heritability and correlation between carcass traits in Japanese Black cattle evaluated under different ceilings of relatedness among individuals[J].Front Genet,2023,14:1053291.
[68] TUSELL L,GILBERT H,VITEZICA Z G,et al.Dissecting total genetic variance into additive and dominance components of purebred and crossbred pig traits[J].Animal,2019,13(11):2429-2439.
[69] WU Z Y,DOU T F,BAI L Y,et al.Genomic prediction and genome-wide association studies for additive and dominance effects for body composition traits using 50 K and imputed high-density SNP genotypes in Yunong-black pigs[J].J Anim Breed Genet,2023,doi:10.1111/jbg.12830.
[70] HAYES B J,COPLEY J,DODD E,et al.Multi-breed genomic evaluation for tropical beef cattle when no pedigree information is available[J].Genet Sel Evol,2023,55(1):71.
[71] SUTERA A M,TOLONE M,MASTRANGELO S,et al.Detection of genomic regions underlying milk production traits in Valle del Belice dairy sheep using regional heritability mapping[J].J Anim Breed Genet,2021,138(5):552-561.
[72] MOGHADDAR N,BROWN D J,SWAN A A,et al.Genomic prediction in a numerically small breed population using prioritized genetic markers from whole-genome sequence data[J].J Anim Breed Genet,2021,139(1):71-83.
(编辑 郭云雁)
猜你喜欢
现代经济信息(2016年19期)2016-10-20 18:46:44
现代经济信息(2016年19期)2016-10-20 18:12:28
现代经济信息(2016年19期)2016-10-20 16:20:30
中国科技博览(2016年19期)2016-10-19 13:33:22
中国科技博览(2016年18期)2016-10-19 10:49:54
中国科技博览(2016年18期)2016-10-19 08:16:45
中国科技博览(2016年18期)2016-10-19 06:39:44
中国市场(2016年36期)2016-10-19 03:54:01
中国市场(2016年35期)2016-10-19 02:30:10
商(2016年27期)2016-10-17 07:09:07
