
4例罕见基因位点突变的新生儿先天性肌病临床表现及基因表型
2024-05-15 09:52:42叶超群代青梅邓璐瑶张语黄会芝
山东医药 2024年12期
叶超群,代青梅,邓璐瑶,张语,黄会芝
安徽医科大学附属省儿童医院新生儿科 安徽医科大学第五临床医学院,合肥 230051
先天性肌病(CM)是一组具有临床、组织病理学和遗传异质性的罕见遗传性肌肉疾病,主要由肌肉纤维的结构异常导致,临床表现为不同程度的肌张力低下和肌力减低、无力,临床病程常呈静止或进展缓慢。CM 的诊断既往主要依靠其特征性肌肉病理改变,并根据病理特征分为5个亚型。由于CM 肌肉活检创伤性较大,医疗单位操作难度大,家长依从性差,且临床类型繁杂,临床表现具有异质性,因此基因表型分析对CM 的临床诊断更具指导意义。随着二代基因测序技术的发展,不断有大量新基因位点突变被确定为先天性肌病的致病基因,拓宽了先天性肌病的表型—基因型谱,为早期发现家系中的潜在遗传基因提供方向并为患儿预后提供线索。本研究回顾性分析了4 例CM 患儿的临床资料及基因检测结果,总结其临床表现及基因表型,为罕见基因位点突变致新生儿CM的诊断提供参考依据。
1 资料分析
病例1:①病历资料:患儿女,出生1 d,因“反应差伴间断抽搐1 天余”入院,患儿系G1P1,出生胎龄39+1周,顺产,出生体质量3 830 g,病程中精神反应差,有青紫、抽搐,人工喂养、吃奶欠佳。入院查体:呼吸60 次/分,心率156 次/分,体质量3 620 g,身长54 cm,头围37 cm,意识不清,反应极差,哭声弱,四肢动作少,压眶反射弱,全身皮肤可见轻中度黄染,前囟平软,双瞳孔等大等圆,对光反射弱,双肺呼吸音粗,双肺可闻及痰鸣音;心音有力,律齐,未闻及病理性杂音,腹软,肠鸣音减弱,四肢肌张力减低,伴有间断抽搐发作,觅食反射减弱,吸吮反射减弱,拥抱反射减弱,握持反射消失,手指、脚趾细长。辅助检查:血清肌酸肌酶(CK)轻度升高380 IU/L;超敏肌钙蛋白I 轻度升高:2.218 ng/mL;心电图示:窦性心动过速;头颅CT提示:脑白质密度减低,蛛网膜下腔出血;双侧枕部硬膜下出血;双侧顶枕部头皮血肿。入院后予禁食、补液、镇静止惊、抗感染等支持治疗,入院第2天出现频繁抽搐,气促伴呼吸困难,心率增快200~204 次/分,全身皮肤可见散在花斑纹,四肢末梢凉,CRT 5 s,血压101/82 mmHg,考虑休克,予气管插管、纠酸、扩容、强心治疗后患儿仍旧处于昏迷,瞳孔对光反射消失,入院第3天家长放弃治疗后出院,出院第5 天患儿在我院急症因呼吸衰竭抢救无效死亡。患儿父母体健,非近亲结婚,无遗传病史。②基因测序结果:患儿存在COL12A1基因c.7622(exon49)C>T(胞嘧啶>胸腺嘧啶)的错义突变,导致氨基酸改变p. S2541F。结合临床表现,患儿诊断为远端肌病4型,遗传方式为常染色体显性遗传。Sanger测序验证提示该患儿表现为杂合突变,该变异先证者为杂合子,受检者父、母亲均为野生型。以本例患者为线索追踪调查,家系中已婚者均为非近亲结婚。该突变点在普通人携带频率数据库如千人基因组、gnomAD等中未收录,为罕见变异。根据美国医学遗传学与基因组学学会(ACMG)分类指南[1]该变异评级为可能致病变异,其致病性依据为PS2+PM2+PP3。
病例2:①病历资料:患儿,女,出生16 d,因“呼吸费力,不能离氧13 d”入院,患儿系G3P3,出生胎龄39+2周,剥宫产,出生体质量4 600 g,Apgar 评分1 min 10分,5 min 10分,生后即出现吃奶费力,哭声不畅,伴有哭闹后口唇。颜面部青紫。入院查体:呼吸60 次/分,心率145 次/分,体质量4 150 g,身长48 cm,头围38 cm,前囟平软未闭,意识清楚,反应一般,哭声不畅,耳位低,鼻梁低平,高腭弓,口唇发绀,颈短,双肺呼吸粗,可闻及较多痰鸣音,心音有力,律齐,未闻及明显病理性杂音,腹软,肠鸣音正常,四肢肌张力稍减低,觅食反射减弱;吸吮反射减弱;拥抱反射:减弱;四肢短小,双手指关节挛缩(左侧第3指、右手2~5指明显);双足小,足趾头短小,呈马蹄内翻足。辅助检查:血清CK 轻度升高114 IU/L;心脏彩超提示:房间隔多发缺损(EF72%,FS38%);耳声发射示:左耳未通过;纤维支气管镜示:先天性喉喘鸣,会厌软化;喉软化I 型,气管支气管黏膜炎症,气管中下段轻度软化;遗传代谢病血、尿筛查:未发现特异性改变;入院后予鼻导管吸氧、雾化、吸痰等对症支持治疗后于第4 天出院,出院后患儿症状逐渐加重,多次因吞咽困难差,呼吸道分泌物较多导致呼吸困难及重症肺炎于我院住院支持治疗,7 月龄时患儿双上肢肌力4级,双下肢肌力3级,肌张力低,并因呼吸衰竭死亡。预后:死亡。患儿父体健,母亲有家族性糖尿病,非近亲结婚。母孕期胰岛素治疗,孕期胎儿染色体及羊水穿刺未见异常。G1男孩,11岁,体健;G2,女孩,4岁,体健。②基因测序结果:患儿存在NEB 基因c.25288C>T(p.Arg8430)的无义突变及c.2783delC(p.Pro928fs)移码突变,导致蛋白质发生截短从而丧失其正常功能。结合临床表现,患儿诊断为杆状体肌病2 型,遗传方式为常染色体隐性遗传。Sanger测序验证提示该患儿表现为杂合突变,该变异先证者为杂合子,受检者父、母亲均为野生型。以本例患者为线索追踪调查,家系中已婚者均为非近亲结婚。该变异在普通人携带频率数据库如千人基因组中未收录,dbSNP147 数据库有收录。ACMG变异评级为疑似致病性变异。
病例3:①病历资料:患儿,女,出生21 h,因“生后口吐泡沫1 h”入院,患儿系G2P2,出生胎龄39+3周,剖宫产,出生体质量2 500 g,Apgar 评分1 min 10分,5 min 10 分。入院查体:呼吸52 次/分,心率125次/分,体质量2 430 g,身长47 cm,头围33 cm,前囟平软,神情,反应好,哭声响亮,双侧外耳廓畸形,耳廓形态异常伴有肉赘,上腭可见约1.5 cm 缺损区,哭时口角向左下歪斜,舌后赘,下颌小,胸骨下端凹陷,双肺呼吸粗,未闻及干湿性啰音,心音有力,律齐,未闻及明显病理性杂音,腹软,肠鸣音正常,四肢肌张力偏高,原始反射可引出。辅助检查:血清CK轻度升高292 IU/L;心电图示:窦性心动过速;心脏彩超提示:1、房间隔缺损;2、动脉导管未闭;3、肺动脉高压;头颅MRI:脑实质未见明显异常信号,硬腭部分缺如,右后鼻道部分狭窄,舌根后赘,上气道窄,两额叶体积稍小;胸部CT示:支气管肺炎;气管支气管畸形可能;心影增大;胸骨远端向内凹陷,漏斗胸;纤维支气管镜检查示:1. 软腭缺如;2. 舌根后赘;3.腺样体肿大;4. 气管支气管黏膜严重;5. 气管源性支气管;听性脑干反应:左耳未通过,右耳未通过。肾脏彩超:左肾肾盂分离,预后:患儿于2 岁时先后行腭裂修补术、左侧副耳切除术及双侧下颌骨重建术。现患儿3 岁,患儿父母体健,非近亲结婚,无遗传病史,G1,男孩,15岁,体健。②基因测序结果:患儿存在FLNC 基因c.4021(exon23)C>T的错义突变,导致氨基酸改变p.R1341X,1385。结合临床表现,患儿诊断为杆状体肌病2 型,遗传方式为常染色体显性遗传。Sanger测序验证提示该患儿表现为杂合突变,该变异先证者为杂合子,受检者父、母亲均为野生型。以本例患者为线索追踪调查,家系中已婚者均为非近亲结婚。该变异在普通人携带频率数据库如千人基因组、gnomAD 等中未收录,为罕见变异。ACMG 变异评级为致病性变异,致病等级PVS1+PS2+PM2+PP5+PP3。
病例4:①病历资料:患儿,女,出生13 h,因“哭声不畅、青紫、反应差13 h”入院,患儿系G2P2,出生胎龄39+5周,剖宫产,出生体质量3 140 g,Apgar评分1 min 8 分,5 min 5 分,10 min 10 分,脐带绕颈2 周,生后即出现哭声弱、呻吟。入院查体:呼吸50次/分,心率145 次/分,体质量3 150 g,身长49 cm,头围34.5 cm,前囟平软,精神反应差,哭声弱,四肢动作少,双肺呼吸粗,可闻及明显湿啰音,心音有力,律齐,可闻及Ⅱ~Ⅲ/6 级杂音,腹软,肠鸣音正常,觅食反射:减弱;吸吮反射:减弱;握持反射:减弱;拥抱反射:减弱;四肢肌张力低,肌力Ⅰ级。辅助检查:血清CK 轻度升高440 IU/L;心脏彩超:房间隔多发缺损;纤维支气管镜检查:1. 声带关闭不全,2. 气管支气管粘膜炎症;彩超(新生儿颅腔):1. 双侧脑室内出血;头颅CT示:双侧大脑半球密度不均匀性减低;头部磁共振平扫示:1. 右侧脑室枕角旁点状短T1信号、T1WI双侧内囊后肢高信号消失:考虑脑损伤;2.右侧脑室枕角略著。髋关节彩超:双侧髋关节发育中,左髋发育不成熟(Graf IIb 型),右髋未见明显脱位(Graf Ⅰ型),双下肢磁共振:双下肢诸骨未见明显异常信号,周围肌肉组织及脂肪组织未见异常。振幅整合脑电图:轻度异常;食道造影提示:会厌功能不全。骨骼肌及心肌HE 染色提示:肌纤维间隙增宽,肌纤维和细胞核大小不同,部分细胞核缩缩消失。部分肌纤维末端由圆柱形变为细长,电镜下表现为肌原纤维排列紊乱,Z 线断裂消失。入院后予呼吸机辅助呼吸,抗感染、补液,鼻饲及静脉营养等支持治疗,患儿于入院55 天后家长放弃治疗出院,出院第二天因呼吸衰竭死亡。患儿父母体健,非近亲结婚,无遗传病史,G1,男孩,2 岁,体健。②基因检测结果:患儿存在UNC45B 基因的2 个变异c.2591(exon20)A>C,c.2357(exon18)T>A 的错义突变,导致氨基酸改变p.H864P;p.M786K。结合临床表现,患儿诊断为肌原纤维肌病11 型,遗传方式为常染色体隐性遗传。Sanger测序验证提示该患儿表现为杂合突变,该变异先证者为杂合子,受检者父亲在变异位点c. 2591(exon20)A>C 中为野生型,母亲及哥哥则为杂合子;受检者父亲在变异位点c.2357(exon18)T>A 中为杂合子,母亲及哥哥则为野生型。以本例患者为线索追踪调查,家系中已婚者均为非近亲结婚。该变异在普通人携带频率数据库如千人基因组、gnomAD 等中未收录,为罕见变异。ACMG变异评级为意义不明变异,其致病性依据PM2+PP3。4例患儿全外显子测序结果见图1。
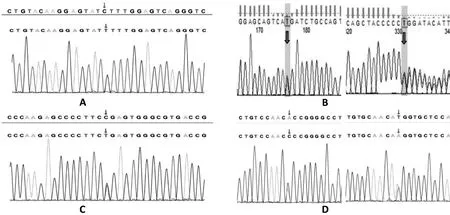
图1 4例患儿基因测序结果
3 讨论
CM 是一组罕见的遗传性原发性肌肉疾病,多在出生时或新生儿期引起肌张力减低和无力,主要累及四肢近端肌肉,当出现面部性肌病时,表现为上睑下垂、眼球下垂及面部畸形如:长脸。小下颌、高腭弓等,骨骼肌受累出现骨骼畸形如:脊柱侧弯、漏斗胸、髋关节脱位或内翻足,严重时会出现心肌及呼吸肌受累,危及生命;智力通常不受影响。CM 的诊断既往主要依靠其特征性肌肉病理改变,并根据病理特征将其分为核心型肌病、线状肌病、中心核型肌病、先天性纤维型失衡肌病、肌球蛋白沉积性肌病[2-3]。但由于肌肉活检创伤性较大,医疗单位操作难度大,家长依从性差,且CM 的临床类型繁杂,临床表现具有异质性,基因表型则对CM 的临床诊断更具指导意义。本文所研究的4 例CM 系通过基因检测明确诊断,分析发现4 种罕见新发位点导致的基因突变,均为新生儿期发病,临床症状较重,主要表现为明显的呼吸困难,原始反射减弱,无家族史,发病早,预后差,死亡率高。提示生后早期出现呼吸困难及肌力进行性减低的患儿往往预后较差。
COL12A1 基因位于染色体6q12-q13,编码XII型胶原蛋白的α链,主要参与纤维形成,参与调节胶原纤维的间距和组装[4],基因突变患者则表现为EDS 综合征样症状,如远端关节活动过度合并近端关节挛缩,异常瘢痕形成,以及肌肉张力降低和肌肉无力伴运动发育迟缓等肌病特征,是一种缓慢进展的罕见CM[5]。NEB基因编码蛋白的分子量在600~900 kDa,可调节肌动蛋白丝的长度。NEB的致病变异是导致杆状体肌病全球范围内最常见的原因之一。突变类型以错义变异为主,可导致早发性远端肌病,主要表现为足部伸肌无力和后来的手部伸肌无力。进展非常缓慢,很少观察到心脏受累,常伴有呼吸问题,其中超过一半的患者需要夜间无创通气。心肌病、上睑下垂和眼肌麻痹在NEB 相关的杆状体肌病患者中很少见,成年患者没有重大残疾[6-7]。FLNC 基因定位于染色体7q32-35,有两个主要转录本,其中长转录本在心肌应激时表达丰富,异常变异时表现为心肌病,短转录本主要在正常情况下表达,且在骨骼肌中的表达量是心肌的3.5倍,突变的典型表现为自成年早期出现的手部及小腿的肌肉无力,心肌同时受累则极为罕见[8]。UNC45 基因有两种亚型,其中UNC-45A 在非肌细胞中广泛表达,通常在肿瘤细胞中升高,被认为有助于其增殖和转移。UNC45B 基因是编码Ⅱ型肌球蛋白折叠所需的伴侣蛋白,参与Ⅱ型肌球蛋白的组装和功能维持,促进横纹肌的收缩。临床表型为早发、缓慢进行性肌无力,以轴向、近端肌无力和呼吸受累为主[9-10]。本文共检索收集该四种基因表型临床病例文献23篇,共报道COL12A1 基因突变致Bethlem 肌病2 型7篇报道共13例患者[11-17],其中男性8例,女性5例,新生儿期发病6例,儿童期发病5例,成人2例,面部畸形4 例,吞咽困难2 例,脊柱畸形3 例,关节受累11例,此外还有一些特异性表现,1 例出现肩关节脱位,4 例出现瘢痕增生;NEB 基因突变致杆状体肌病2型8篇报道共21例患者[18-25],男性9例,女性11例,性别未知1例,新生儿期发病3例,儿童期发病3例,成人10 例,流产6 例,呼吸困难3 例,面部畸形4 例,脊柱畸形2 例,低眼压1 例;FLNC 基因突变致远端肌病4 型家系6 篇共13 例患者[26],女性6 例,男性7例,儿童2例,成人11例,出现呼吸困难1例,心肌受累3 例,其中1 例为心肌和呼吸肌同时受累;UNC45B 基因突变致肌原纤维肌病11 型2 篇报道共12例患者[9],其中男性8例,女性3例,新生儿期发病1 例,儿童期发病10 例,均有运动发育迟滞表现,CK正常或轻度升高,出现呼吸困难1例,面部畸形1例,吞咽困难3例,关节受累2例。
结合国内外病例,FLNC、UNC45B、NEB、COL12A1基因突变相关的肌病都系常染色体遗传疾病,临床表现各异,这些肌病多数在儿童及成年期发病,新生儿期即有明显症状者罕见,多表现为进行性肌无力,部分病人无明显临床表现,仅表现为轻度肌力异常,重症则伴有呼吸肌受累,并有严重的肌张力低下,但各病例患者的CK 均无明显增高。其中有6例患儿为孕早期流产。治疗上,目前还没有可以治愈CM 的方法,只能给予对症治疗,如关节挛缩及脊柱侧弯者给予矫形治疗;吞咽障碍和喂养困难予吞咽训练或鼻饲、甚至胃造瘘术以保证充足的营养支持;呼吸功能障碍者予呼吸训练、肺部物理治疗,必要时可给予机械通气支持。诊断上,随着二代基因测序的发展,全外显子测序及全基因组测序逐渐发展,使得CM患者的检查顺序正在改变,在已知基因致病突变的情况下,或将不再需要通过有创的肌肉活检进行组织病理学检查来对患儿进行临床诊断及分型,可见遗传评估优于肌肉活检,应在诊断过程中首先考虑。此外,基因诊断可以为患者家庭的遗传咨询提供帮助;孕早期的基因测序可以辅助临床筛选出遗传性疾病并进行一级预防,为父母提供可以接受的生育选择,如人工流产及选择性绝育,并通过孕早期健康教育,降低下一胎及下一代患相同疾病的风险。
总之,罕见基因位点突变致新生儿CM 临床表现均有异质性,表现为肌张力及肌力的严重降低,并伴有不同程度的并发症,临床症状不典型;分别发现FLNC 基因、UNC45B 基因、NEB 基因、COL12A1基因4个罕见新发位点突变,其中3个错义突变、1个无义突变合并移码突变。
猜你喜欢
新民周刊(2022年27期)2022-08-01 07:04:49
今日农业(2021年5期)2021-11-27 17:22:19
现代畜牧科技(2021年5期)2021-07-20 08:07:40
传染病信息(2021年6期)2021-02-12 01:52:58
趣味(数学)(2020年4期)2020-07-27 01:44:16
支部建设(2020年15期)2020-07-08 12:34:32
兽医导刊(2016年6期)2016-05-17 03:50:27
百科知识(2015年18期)2015-09-10 07:22:44
中国当代医药(2015年26期)2015-03-01 02:07:11
生物医学工程学进展(2015年1期)2015-02-28 14:53:42
