
基因缺失杆状病毒载体的构建及应用
2024-05-09 08:29王同燕仝晓丹苏晓蕊宋欢欢李伟国刘武杰谭菲菲田克恭
中国动物传染病学报 2024年1期
王同燕,仝晓丹,苏晓蕊,宋欢欢,李伟国,刘武杰,谭菲菲,田克恭,3
(1.国家兽用药品工程技术研究中心,洛阳 471003;2.普莱柯生物工程股份有限公司,洛阳 471000;3.洛阳普泰生物技术有限公司,洛阳 471003)
猪瘟(Classical swine fever, CSF)是由猪瘟病毒(Classical swine fever Virus, CSFV)引起的一种急性、热性、高度接触性传染病,给中国和其他国家养猪业造成了严重危害,被世界动物卫生组织列为必须报告的动物疫病之一[1]。虽然中国利用自行研制的猪瘟兔化弱毒疫苗进行强制免疫接种,有效控制了猪瘟的大规模流行。但是活疫苗免疫抗体与野毒感染抗体无法利用血清学方法进行鉴别,成为我国净化CSF面临的关键科学问题[2-3]。因此有必要研制安全、可靠、可区分野毒感染的新型标记猪瘟疫苗。猪瘟E2蛋白位于病毒囊膜表面,参与病毒的感染过程,是CSFV的主要保护性抗原蛋白之一,能诱导机体产生中和抗体[4]。
杆状病毒表达载体系统(Baculovirus expression vector system, BEVS)是利用携带有外源目的基因的重组杆状病毒作载体在昆虫体内或细胞中生产重组蛋白的表达系统。该系统能对外源蛋白进行磷酸化和糖基化等翻译后修饰,使蛋白接近天然构象,保持良好的生物活性和免疫原性,并且利于大量生产,因此杆状病毒系统被广泛用于亚单位疫苗的研发。但杆状病毒表达系统也存在两个主要的缺陷:第一外源蛋白表达量较低,主要原因在于杆状病毒本身携带多个蛋白酶基因,如几丁质酶(Chitinase,ChiA)和组织蛋白酶(viral cathepsin-like protein,V-cath),表达外源基因的同时这些蛋白酶也随之表达,特别是在病毒感染细胞晚期,重组蛋白易受这些蛋白酶降解,使获得的目的蛋白的量减少。另一个原因是p10和pH均属于晚期强启动子,可以启动蛋白高效表达,但是目前常用的Bac-to-Bac系统中两个强启动子在表达外源基因时存在竞争关系,最终影响外源蛋白的表达量。第二是杆状病毒传代效应,即随着杆状病毒传代,出现缺陷干扰颗粒(Defective Interfering particle, DIs),致使外源蛋白表达量下降。其中FP25K和DA26基因中有宿主转座整合位点,转座插入导致DIs产生,影响杆状病毒基因组稳定性和外源蛋白表达量[5-8]。
为了提高杆状病毒表达量和解决杆状病毒毒种代次窄的问题,本研究首次同时敲除ChiA、V-cath、P10、FP25K和DA26等5个影响蛋白表达和基因组稳定性的基因,从而获得表达量提高和基因稳定的杆状病毒载体,猪瘟E2蛋白的表达水平提高约4倍,同时使重组杆状病毒的种毒代次从7代延长至20代左右,解决了杆状病毒表达量和种子批代次范围窄的问题,为开发猪瘟病毒亚单位疫苗提供技术支撑。
1 试验材料及方法
1.1 主要试验材料 pFastBac-Hr1-WPRE载体、pKD46质粒、pCas质粒、pTarget Dual质粒、pPICZaA质粒、pBeloBAC11质粒、DH10Bac感受态细胞均由国家兽用药品工程技术研究中心构建和保存;Sf9细胞、ExpiSf9细胞、High Five(Hi5)细胞均有国家兽用药品工程技术研究中心传代、保存;opti-MEM、ExpiSf CD Medium、Express Five SFM、Sf-900 II SFM培养基和ExpiFectamine Sf转染试剂均购自Thermo公司;猪瘟阳性血清由国家兽用药品工程技术研究中心保存;兔抗猪IgG-HRP购自Sigma公司;His抗体购自MBL公司;His TrapTMHP预装柱购自GE公司;Viral Nucleic Acid Extraction Kit购自Geneaid公司;BacPAKTMBaculovirus Rapid Titer Kit购自Clontech公司。
1.2 引物及sgRNA 根据P10、ChiA和V-cath基因在病毒基因组的位置及基因序列,设计重组缺失引物,其中ChiA和V-cath(CC)基因位置相邻同时缺失(表1)。同时根据Case9技术原理,设计DA26和FP25K缺失所需的sgRNA序列(表2)。
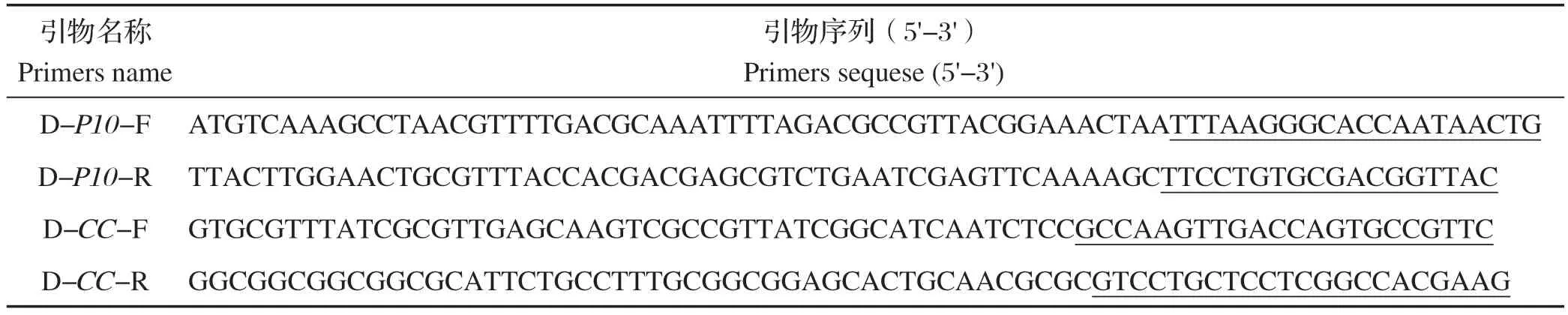
表1 P10 和CC 基因缺失引物Table 1 Deletion of primer for P10 and CC genes
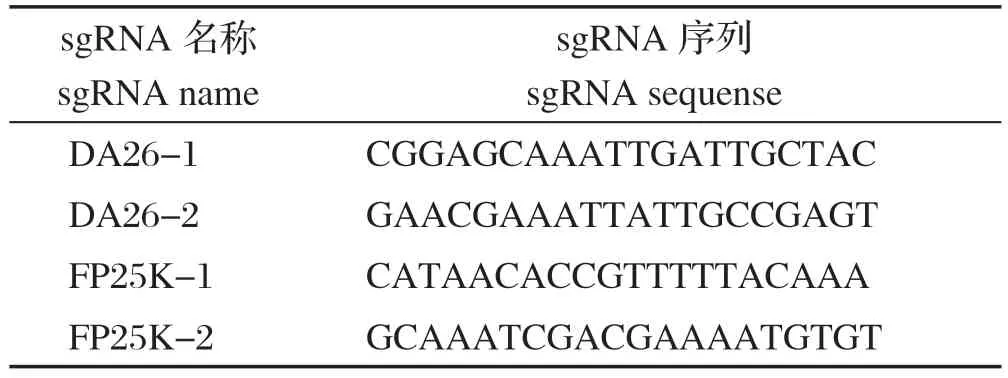
表2 DA26、FP25K 基因缺失sgRNA 序列Table 2 Sequences of sgRNA for DA26 and FP25K genes deletion
1.3ChiA、V-cath、P10、FP25K和DA26五基因缺失菌株的构建 首先利用Red重组技术[9]缺失ChiA、V-cath和P10基因。因ChiA基因和V-cath基因位置相邻,故设计1对重组引物进行两个基因的同时删除,根据ChiA左侧序列和V-cath基因右侧序列设计引物来扩增含有同源臂的博莱霉素基因。将其电转化至含有杆状病毒基因组Bacmid和pKD46的DH10B感受态中,借助博莱霉素抗性进行压力筛选获得缺失ChiA和V-cath(CC)基因的阳性克隆。在缺失ChiA和V-cath基因的基础上,按照同样的方法利用氯霉素基因替换P10基因,获得ChiA、V-cath基因和P10基因缺失的阳性质粒。
在缺失ChiA、V-cath和P10的基础上,利用CRISPR在线设计工具网站http://crispr.mit.edu/,设计FP25K基因和DA26基因的SgRNA序列,分别选取两个基因评分比较高的1对sgRNA,委托生工生物工程(上海)股份有限公司优化合成,构建针对同一基因不同位点的双sgRNA质粒,将此sgRNA质粒和含有Cas9的质粒共转化至已缺失ChiA、V-cath基因和P10基因的DH10Bac感受态中,通过PCR筛选目的基因缺失的阳性质粒。
1.4 E2基因供体质粒构建及鉴定 在线预测CSFV E2蛋白的糖基化位点,同源建模分析CSFV E2蛋白的二聚体结构,将可能影响E2蛋白同源二聚体形成的糖基化位点进行突变,即将E2基因的297位天冬酰胺(N)突变为谷氨酰胺(Q),并在N端添加GP67信号肽。将此E2基因序列委托生工生物工程(上海)股份有限公司优化合成。将优化合成的E2基因通过BamHⅠ和HindⅢ酶切位点插入到pFastBac-Hr1-WPRE改造载体中[10],酶切鉴定正确的供体质粒送金唯智生物科技有限公司进行测序。
1.5 重组Bacmid的构建及鉴定 将含有突变E2基因的供体质粒和含有转座酶的Helper质粒共转化至含有五基因缺失杆状病毒基因组的DH10Bac感受态细胞中,利用Bac-to-Bac系统的转座原件,实现E2基因转座至杆状病毒基因组中,通过两轮蓝白斑筛选,挑选白色菌落于含有卡那霉素、庆大霉素和四环素的三抗液体LB中,37℃震荡培养过夜。按照碱裂解和异丙醇沉淀核酸的方法抽提核酸,利用E2特异性引物E2-F:ATGCTACTAGTAAATCAG和E2-R:TCAGTGATGGTGATGGTGAT鉴定重组Bacmid。同时设置含有突变E2基因的供体质粒转化DH10Bac感受态作为对照。
1.6 重组杆状病毒拯救 将鉴定正确的重组Bacmid按照传统贴壁的方法[11]转染Sf9细胞,待细胞出现明显细胞病变时收获上清液作为P1代重组杆状病毒。同时将鉴定正确的重组Bacmid悬浮转染ExpiSf9细胞。将处于对数生长期的ExpiSf9细胞密度调整为2.5×106cells/mL,取120 μL ExpiFectamineTMSf Transfection Reagent加入1 mL opti-MEM中,颠倒混匀10次,室温静置5 min,加入50 μg重组Bacmid核酸,轻柔混匀10次,室温孵育15 min后,将混合液加入到100 mL细胞密度为2.5×106cells/mL ExpiSf9细胞中,在27℃培养箱中120 r/min震荡培养,待细胞病变达70%~80%时收获培养上清液,标记为P1代重组杆状病毒。参照Clontech公司的BacPAK™Baculovirus Rapid Titer Kit说明书进行重组杆状病毒滴度测定,比较两种转染方式获得的P1代重组病毒的滴度。
1.7 重组杆状病毒鉴定
1.7.1 目的基因鉴定 参照Genaid公司的Viral Nucleic Acid Extraction Kit说明书进行P1代重组杆状病毒基因组抽提,以抽提的核酸为PCR模板,利用E2特异性引物E2-F和E2-R进行重组杆状病毒的基因鉴定。
1.7.2E2基因转录和表达 将收获的P1代重组杆状病毒按照MOI=1接种Hi5细胞,48~72 h收获细胞离心取上清液进行Western blot鉴定和E2蛋白的亲和层析纯化,Western blot分别利用HRP标记-His单克隆抗体(1∶8000)和猪瘟阳性血清(1∶500)进行鉴定。E2蛋白亲和纯化条件为:20 mmol/L Tris-HCl,150 mmol/L NaCl,40 mmol/L咪唑,pH7.5,洗涤杂蛋白,20 mmol/L Tris-HCl,150 mmol/L NaCl,300 mmol/L 咪唑,pH7.5,洗脱目的蛋白。收集洗脱液,进行SDS-PAGE鉴定,分别在还原和非还原条件下分析E2蛋白二聚体形成的情况,并参照BCA试剂盒说明书对洗脱的E2蛋白进行定量。
1.7.3 重组杆状病毒基因组稳定性 将重组杆状病毒按照MOI=0.01接种Sf9细胞,接种后72~96 h收获病毒进行继续传代;取P3、P5、P7、P10、P15、P20和P25代重组杆状病毒按照MOI=1接种Hi5细胞,接种48~72 h收获细胞上清液,Western blot鉴定E2蛋白的表达。
2 结果
2.1 ChiA、V-cath、P10、FP25K和DA26五基因缺失菌株的构建 五基因缺失质粒构建示意图见图1,首先利用Red重组技术分别将Zeocin和CmR替换ChiA/V-cath基因和P10基因,分别在ChiA/V-cath基因外侧和P10基因外侧设计检测引物,通过PCR检测重组阳性质粒;通过Cas9技术,双切FP25K基因和DA26基因,在切口的外侧设计检测引物,通过PCR检测敲除FP25K和DA26的阳性质粒。将PCR检测阳性的样品送测序,测序结果表明五个基因均已成功缺失,获得的五基因缺失阳性菌株命名为rBacmid-5D。

图1 ChiA、V-cath、P10、FP25K 和DA26 五基因缺失示意图Fig.1 Schematic diagram of f ive deletions of ChiA、V-cath、P10、FP25K and DA26
2.2 表达E2蛋白的供体质粒构建及鉴定 利用BamHⅠ和HindⅢ双酶切鉴定构建的供体质粒,结果可见约1100 bp的目的条带,见图2。测序结果和优化合成的序列一致,酶切和测序均正确的质粒命名为pFastBac-Hr1-WPRE-E2。

图2 pFastBac-Hr1-WPRE-E2 酶切鉴定Fig.2 pFastBac-Hr1-WPRE-E2 digestion and identif ication
2.3 重组Bacmid的构建及鉴定 将供体质粒pFastBac-Hr1-WPRE-E2分别转化DH10Bac和rBacmid-5D感受态细胞,对获得的重组Bacmid进行PCR鉴定,可见1100 bp的目的条带,阴性对照未见目的条带(图3)。鉴定正确的Bacmid分别命名为rBacmid-E2和rBacmid-5D-E2。
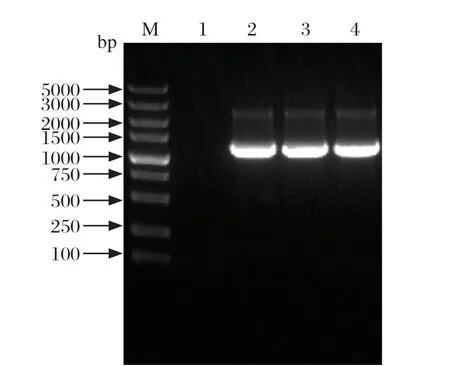
图3 重组Bacmid 的PCR 鉴定Fig.3 PCR identif ication of recombinant Bacmid
2.4 重组杆状病毒拯救及鉴定 将鉴定正确的rBacmid-E2和rBacmid-5D-E2分别贴壁转染Sf9细胞和悬浮转染ExpiSf9细胞,收获上清液即为P1代重组杆状病毒,分别名字为rAc-E2和rAc-5D-E2。贴壁和悬浮转染收获的P1代rAc-E2的病毒滴度分别为1.08×107IFU/mL和7.35×108IFU/mL。贴壁和悬浮转染收获的P1代rAc-5D-E2的病毒滴度分别为1.12×107IFU/mL和8.01×108IFU/mL。
2.4.1 目的基因鉴定 抽提悬浮转染获得的重组杆状病毒核酸进行PCR鉴定,拯救的两种重组杆状病毒均能扩增出约1100 bp特异条带(图4),测序结果表明插入的E2基因序列完全正确。

图4 E2 基因鉴定Fig.4 E2 gene identif ication
2.4.2 E2基因转录和表达 将悬浮转染获得的两种重组杆状病毒rAC-E2和rAC-5D-E2按照MOI=1接种Hi5细胞,接种48 h上清液中可检测到和His抗体以及猪瘟阳性血清均发生反应的特异性条带,条带大小约50 kDa(图5)。对收获上清液进行亲和层析纯化,纯化获得的E2蛋白纯度均在95%以上,而且在非还原条件下分析E2蛋白95%以上为二聚体形式(图6)。采用BCA的方法对rAC-E2和rAC-5D-E2表达E2蛋白进行定量,分别为35 mg/L和138 mg/L。

图5 Western blot 鉴定Fig. 5 Western blot identif ication
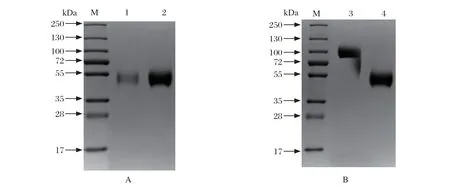
图6 SDS-PAGE 鉴定Fig.6 SDS-PAGE identif ication
2.4.3 重组杆状病毒基因组稳定性 将重组杆状病毒rAC-5D-E2和rAC-E2分别按照MOI=0.01接种Sf9细胞,接种后72~96 h收获病毒,并继续进行传代。取P3、P5、P7、P10、P15、P20和P25代重组杆状病毒按照MOI=1接种Hi5细胞,接种48 h收获细胞上清液,进行Western blot鉴定,结果rAC-5D-E2 P20代内E2的蛋白量基本保持不变;而rAC-E2从P7代开始表达量明显降低(图7)。

图7 不同代次重组杆状病毒表达E2 蛋白的Western blot 鉴定Fig.7 Western blot identif ication of E2 protein expressed by diff erent passages of recombinant baculovirus
3 讨论
鉴于猪瘟对养猪业造成的严重危害,开发安全、可靠,同时又可鉴别野毒感染的亚单位疫苗尤为重要。研究报道E2亚单位疫苗具有安全性高、可进行鉴别诊断等优点,是新型疫苗的重点研究方向[12-14],猪瘟E2蛋白位于病毒囊膜表面,参与病毒的感染过程,E2蛋白可以和E1蛋白形成异源二聚体,也可以自身形成同源二聚体,同源二聚体能诱导机体产生中和抗体,是CSFV的主要保护性抗原蛋白。猪瘟病毒C株E2蛋白胞外区含有15个半胱氨酸,其中前14个形成分子内二硫键,294位半胱氨酸参与分子间二硫键的形成;E2蛋白胞外域中存在7个潜在的糖基化位点,其中297位糖基化位点可能影响分子间二硫键的形成[15],在优化序列合成时对297位天冬酰胺(N)突变为谷氨酰胺(Q),表达的E2蛋白95%以二聚体形式存在。
杆状病毒表达系统具有容量大、蛋白翻译后修饰等诸多优势,已被成功应用于亚单位开发和蛋白结构功能研究。但面对产业化,仍存在生产成本较为昂贵的缺点[16];其次是杆状病毒感染后蛋白表达处于晚期,此时细胞面临崩解的状态,致使靶蛋白的转录表达水平和翻译后修饰均未达到最佳状态[17];同时存在随着重组杆状病毒传代,基因组不稳定存在插入和丢失现象,外源基因也会发生丢失。杆状病毒表达系统存在的问题,在一定程度限制了BEVS在产业化方面的应用。为了克服BEVS存在的不足,Lee等[18]通过缺失几丁质酶和组织蛋白酶,使纤维素酶的表达量提高17%。Kim等[19]通过RNAi技术抑制组织蛋白酶的表达,提高GFP蛋白的表达量3倍以上。李国辉等[20]通过缺失几丁质酶和半胱氨酸蛋白酶基因,延长病毒感染的昆虫细胞存活时间,抑制靶蛋白的降解,提高NS1的表达量达3倍。本研究通过Red重组技术和Cas9技术,针对影响杆状病毒表达水平和基因稳定的ChiA、V-cath、P10、FP25K和DA26基因同时缺失,利用五基因缺失后的杆状病毒载体构建的表达猪瘟E2蛋白重组杆状病毒的转录水平和表达水平均有明显提高。本文同时比较了用于拯救重组杆状病毒的两种不同的转染方法,表明悬浮转染不仅获得足量的低代次病毒,而且病毒滴度是传统转染方法获得病毒滴度的70倍左右。因为P1代拯救重组杆状病毒滴度较高,无需通过扩繁病毒即可进行蛋白表达,缩短了从构建到获得蛋白的时间。
综上,本研究通过对重组杆状病毒基因组进行了独创的五基因缺失,使杆状病毒基因组更稳定,同时采用悬浮转染获得了足量低代次高滴度的重组杆状病毒,从而使得表达E2代的重组杆状病毒毒种从P7代延长至P20代,解决了产业化过程中重组杆状病毒种子批代次窄的问题。本文通过对目的基因序列的优化设计、载体杆状病毒基因组的改造以及悬浮转染技术,有效地提高了目的蛋白的表达量,并提高了重组杆状病毒的传代稳定性,为其他杆状病毒源亚单位疫苗的开发提供了一条可行的技术路径。
猜你喜欢
山东畜牧兽医(2022年10期)2022-12-06
当代水产(2021年7期)2021-11-04
今日农业(2021年11期)2021-08-13
幸福(2019年12期)2019-05-16
猪业科学(2018年8期)2018-09-28
中国兽医杂志(2016年5期)2016-06-27
兽医导刊(2016年6期)2016-05-17
广东海洋大学学报(2015年4期)2016-01-13
遗传(2014年3期)2014-02-28
世界科学(2014年8期)2014-02-28
