
Construction and validation of somatic mutation-derived long noncoding RNAs signatures of genomic instability to predict prognosis of hepatocellular carcinoma
2024-05-07 13:21BoTaoDuanXueKaiZhaoYangYangCuiDeZhengLiuLinWangLeiZhouXingYuanZhang
Bo-Tao Duan,Xue-Kai Zhao,Yang-Yang Cui,De-Zheng Liu,Lin Wang,Lei Zhou,Xing-Yuan Zhang
Abstract BACKGROUND Long non-coding RNAs (LncRNAs) have been found to be a potential prognostic factor for cancers,including hepatocellular carcinoma (HCC).Some LncRNAs have been confirmed as potential indicators to quantify genomic instability (GI).Nevertheless,GI-LncRNAs remain largely unexplored.This study established a GI-derived LncRNA signature (GILncSig) that can predict the prognosis of HCC patients.AIM To establish a GILncSig that can predict the prognosis of HCC patients.METHODS Identification of GI-LncRNAs was conducted by combining LncRNA expression and somatic mutation profiles.The GI-LncRNAs were then analyzed for functional enrichment.The GILncSig was established in the training set by Cox regression analysis,and its predictive ability was verified in the testing set and TCGA set.In addition,we explored the effects of the GILncSig and TP53 on prognosis.RESULTS A total of 88 GI-LncRNAs were found,and functional enrichment analysis showed that their functions were mainly involved in small molecule metabolism and GI.The GILncSig was constructed by 5 LncRNAs (miR210HG,AC016735.1,AC116351.1,AC010643.1,LUCAT1).In the training set,the prognosis of high-risk patients was significantly worse than that of low-risk patients,and similar results were verified in the testing set and TCGA set.Multivariate Cox regression analysis and stratified analysis confirmed that the GILncSig could be used as an independent prognostic factor.Receiver operating characteristic curve analysis of the GILncSig showed that the area under the curve (0.773) was higher than the two LncRNA signatures published recently.Furthermore,the GILncSig may have a better predictive performance than TP53 mutation status alone.CONCLUSION We established a GILncSig that can predict the prognosis of HCC patients,which will help to guide prognostic evaluation and treatment decisions.
Key Words: Genomic instability;Long noncoding RNA;Hepatocellular carcinoma;Prognosis;Diagnosis
lNTRODUCTlON
Hepatocellular carcinoma (HCC) is one of the cancers with the highest mortality rate among all malignant tumors,ranking sixth among common cancers[1,2].Worldwide,the mortality rate of patients with HCC ranks second among the total mortality of all cancers.The incidence rate and mortality of liver cancer in East Asia,Southeast Asia,Africa and southern Europe are particularly prevalent[3].The incidence rate of HCC is increasing year by year,which is,of course,related to the improvement in diagnostic mode and the shortening of cancer monitoring interval.In the past,viral hepatitis was the main epidemiological cause of HCC,but with implementation of hepatitis B vaccination and the hepatitis C treatment plan worldwide,the annual incidence rate of HCC with viral hepatitis as the main cause has decreased.In addition,increasing evidence suggests that non-alcoholic fatty liver disease and non-alcoholic steatohepatitis (NASH) contribute to the development of HCC and are becoming increasingly common causes of HCC worldwide.With the implementation of viral hepatitis treatment plans,the epidemiological etiology of HCC is likely to shift from viral hepatitis to NASH[4-6].It is well known that HCC is a fairly complex disease.The current prognostic factors for HCC include tumor size,number,vascular invasion,extrahepatic spread,severity of underlying liver disease as defined by bilirubin and portal hypertension,as well as corresponding qualified treatment modalities[7].Traditional surgical treatment and locoregional therapies have obvious efficacy for some HCC patients,but some patients still have the possibility of long-term recurrence,with poor prognosis and high mortality[8].In systematic treatment regimens,advanced HCC patients can generally be treated with tyrosine kinase inhibitors (TKIs).With the increasing understanding and characterization of the immune characteristics of the tumor microenvironment,immune checkpoint inhibitors (ICI) methods further expand the systemic treatment of HCC.The current emerging comprehensive systemic treatment method combines the above two methods,and there is evidence that the combination therapy of ICI+TKI has achieved certain results.However,existing research evidence suggests that the treatment options currently used in clinical practice are still relatively ineffective.In fact,although the efficacy has significantly improved following the introduction of ICI,the objective response rate to treatment is still largely inadequate.Most patients do not have good responses,and the 5-year overall survival (OS) of metastatic HCC is still unsatisfactory.Currently,efforts should mainly focus on expanding treatment targets and searching for reliable biomarkers as much as possible,which will help adjust treatment choices and avoid the risks and costs associated with drug ineffectiveness and side effects[9].Therefore,new biomarkers are eagerly needed to predict the prognosis of HCC patients.
Genomic instability (GI) has been verified to be one of the characteristics of malignant tumors[10].Chromosomal instability and microsatellite instability are two major types of GI,and more importantly,they are significantly associated with the prognosis of cancer patients[11].The underlying mechanism may be related to the oxidative stress response and the joint defect of DNA damage checkpoint and repair pathway[12].It also proves that molecular markers have great potential in quantifying GI.For example,Mettuet al[13] demonstrated that their identified 12-gene GI signature could predict disease outcomes in multiple cancer types with epithelial origins.A mutation-derived gene signature of GI that can help in predicting the OS of patients with HCC was constructed by Songet al[14].Therefore,these GI signatures may be a potential new therapeutic direction for HCC patients.
Long non-coding RNAs (LncRNAs) are non-protein coding transcripts greater than or equal to 200 nucleotides in length[15].More and more evidence suggests that LncRNA is becoming a potential regulator for GI and to some extent quantifying the level of GI[16,17].For example,some studies have found that a discovered NORAD or LINC00657 regulates genomic stability by isolating pumilio proteins[18].LncRNA dysfunction is closely associated with the occurrence of various tumors,including HCC[19].Liet al[20] found that LncRNA Ftx overexpression promoted the proliferation,invasion and migration of HCC cells[20].Although a considerable number of LncRNAs have been discovered to be related to genomic stability,the clinical application of other GI-LncRNAs in cancer has largely been unexplored,but have great potential as new prognostic biomarkers.
Therefore,in our study,we attempted to establish a GI-derived LncRNA signature (GILncSig) that could help predict the prognosis of HCC patients by combining the LncRNA expression profile with the somatic mutation profile.
MATERlALS AND METHODS
Data sources
The data in this study mainly included clinical characteristics,somatic mutation information,and transcriptome expression data of HCC which were extracted from TCGA portal (https://portal.gdc.cancer.gov/).A total of 424 files with mRNA and LncRNA profiles (including 50 normal and 374 tumor tissues),377 clinical characteristics of HCC patients and 372 patients with somatic mutation information were obtained.All HCC patients (n=343) were randomly divided into the training set and the testing set (chi-square test showed that there was no statistical difference between the training set and the testing set) for further construction and verification of the LncRNA signature.
Identification of GI-LncRNAs
In order to identify GI-LncRNAs,we first calculated the cumulative number of somatic mutations for each patient in HCC samples by combining the LncRNA expression profile and the somatic mutation profile,and arranged them from large to small.The patients in the top quarter are referred to as genomically unstable (GU) samples,and the patients in the bottom quarter are genomically stable (GS) samples.The differentially expressed LncRNAs [absolute value of fold change was greater than 1,and the adjustedPvalue of false discovery rate (FDR) was less than 0.05] between the two groups were defined as genome instability-associated LncRNAs.
Hierarchical cluster analysis was performed on all samples,and differentially expressed LncRNAs were used to identify the GU-like group and GS-like group.In order to examine the correlation between GI-LncRNAs and mRNA pairings,the top 10 mRNAs most related to each GI-LncRNA were screened using the Pearson correlation coefficient.On this basis,a co-expression network was established.Subsequently,functional enrichment analysis was performed on the co-expressed LncRNA-associated mRNAs to reveal the potential biological characteristics of GI-LncRNA,including Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways.ClusterProfiler software in Rversion 4.0.2[21] was used for functional enrichment analysis.
Establishment of the GILncSig
In the training set,the GILncSig formula with risk score was established based on the results of multivariate Cox regression analysis and the expression level of GI-LncRNA.The formula was as follows: GILncSig (patient)=∑(expression of LncRNAn*coef (LncRNAn)),where GILncSig (patient) is a prognostic risk score for the HCC patient,and the LncRNAn represents the nth of independent prognostic LncRNAs.The coef (LncRNAn) represents the contribution index of LncRNAi to prognostic risk score from the Cox regression analyses[21].In the training set,patients' median risk score was used as a dividing line between patients in the high-risk group (high GILncSig) and those in the low-risk group (low GILncSig).The prediction ability of GILncSig was verified by the Kaplan-Meier (K-M) method (P< 0.05 was considered significant).Moreover,the performance was further evaluated by the time-dependent receiver operating characteristic (ROC) curve.All calculations and analyses in this paper were performed using R-version 4.0.2.
Validation of the GILncSig
We first validated the model on a randomly assigned test set and a TCGA set containing all patients.Similar to the training set,we used the GILncSig to calculate the risk scores of each patient within the two sets separately and divided them into two groups of high and low risk within the respective sets.The same K-M analysis and ROC curves were used to validate the GILncSig between the two groups in each of the two pools.Secondly,we used Cox regression analysis to verify whether the GILncSig could be distinguished from other clinical features as an independent prognostic factor.We also performed ROC curve analysis of the GILncSig with two extant LncRNA signatures predicting HCC prognosis and compared their area under the curve (AUC),and then we verified whether the GILncSig could be applied to patients with different clinical characteristics using K-M analysis.In addition,we also analyzed the prognostic value of the GILncSig in combination with TP53.
RESULTS
Identification of GI-LncRNAs in HCC
According to the number of somatic mutations in each patient,we were able to establish the GS group (n=90) and GU group (n=93).Differential expression analysis of LncRNA expression profiles of the two groups was then conducted,and 88 different LncRNAs with statistical significance were obtained (|fold change| > 1 and FDR adjustedP< 0.05).Of these,56 LncRNAs were found to be upregulated and 32 to be downregulated.The heat map (Figure 1A) shows the top 20 LncRNAs with the largest differential expression.Unsupervised hierarchical clustering analysis was performed on all HCC samples based on the expression levels of 88 differentially expressed LncRNAs,and 374 samples were divided into two groups,which are shown in Figure 1B.The lever of somatic mutations in the GU-like group was significantly higher than in the GS-like group (Figure 1C).In addition,the expression ofH2AXwas compared between the two groups.It was found that the expression ofH2AXin the GU-like group was significantly higher than that in the GS-like group (P< 0.01,Figure 1D).H2AXhas been found to promote rapid division of cancer cells and is significantly associated with GI[22].
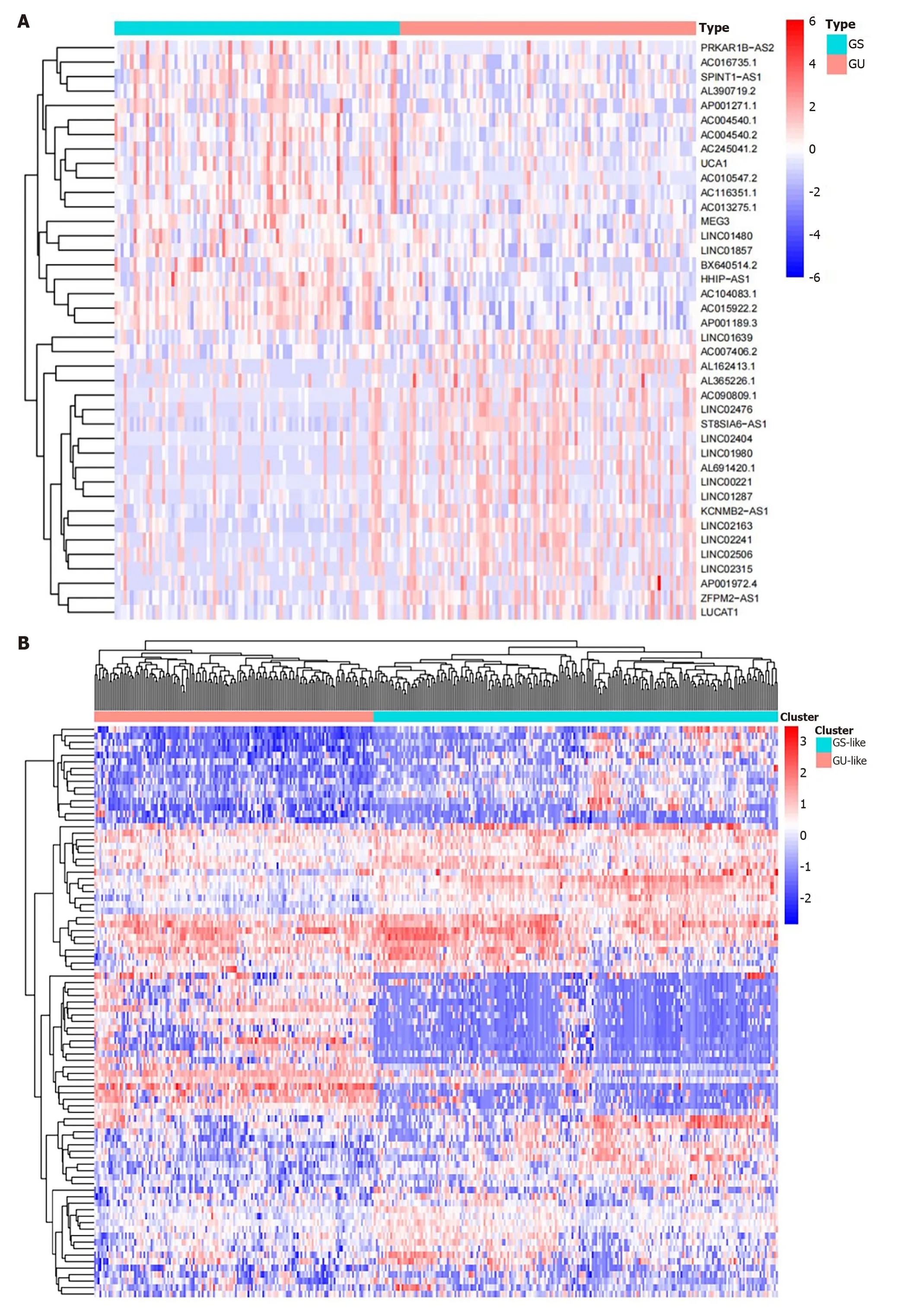
Figure 1 ldentification of genomic instability-related long non-coding RNAs in patients with hepatocellular carcinoma. A: The top 20 long non-coding RNAs (LncRNAs) significantly expressed between the genomically unstable (GU) and genomically stable (GS) groups;B: Unsupervised hierarchical clustering analysis was conducted on 374 tumor samples in the TCGA set using 88 differentially expressed LncRNAs.The left orange cluster is the GU-like group,and the right blue cluster is the GS-like group;C: Boxplots of somatic mutations between the GU-like group and GS-like group;D: Boxplots of H2AX expression level in the GU-like group and GS-like group.The expression level of H2AX in the GU-like group is significantly higher than that in the GS-like group.GU: Genomically unstable;GS: Genomically stable.
Next,we used functional enrichment analysis to predict the potential functions of these GI-LncRNAs.We screened the top 10 protein-coding genes (PCGs) with the strongest correlation with LncRNA.On this basis,an LncRNA-mRNA coexpression network was constructed (Figure 2A).The GO analysis of co-expressed LncRNA-associated mRNAs showed that mRNAs and LncRNA-corrected PCG in the network were significantly enriched in the metabolic process,including the small molecule catabolic process and fatty acid metabolic process (P< 0.05,Figure 2B).In terms of KEGG pathway analysis,22 significantly rich pathways were found,including pyrimidine metabolism,purine metabolism,and folate biosynthesis (P< 0.05,Figure 2C).The results of functional enrichment analysis showed that 88 differentially expressed LncRNAs could participate in a variety of cancer-related biological processes by interfering with a variety of metabolic pathways,among these processes,gene instability could be affected by interfering with gene synthesis.

Figure 2 Functional analysis of the genomic instability-related long non-coding RNAs. A: Co-expression network of genomic instability-related long non-coding RNAs (LncRNAs) and mRNAs.The red circles represent mRNAs,and the blue circles represent LncRNAs;B and C: Functional enrichment analysis of Gene Ontology terms and Kyoto Encyclopedia of Genes and Genomes for mRNAs co-expressed with LncRNAs.
Development of a GILncSig outcome prediction in the training set
To further explore the prognostic effects of these GI-LncRNAs,we randomly divided all HCC patients into two groups: The training set (n=172) and testing set (n=171).Univariate Cox regression analysis was performed on the samples in the training set to analyze the association between OS and LncRNA expression levels of 88 GI-LncRNAs in the training set.A total of 13 LncRNAs were discovered to be significantly correlated with the prognosis of HCC patients (P< 0.05,Figure 3A).Multivariate Cox regression analysis of these 13 LncRNAs was then conducted.Finally,5 of the 13 candidates LncRNAs (miR210HG,AC016735.1,AC116351.1,AC010643.1andLUCAT1) in multivariate Cox analysis showed prognostic significance,and are considered to be independent prognostic factors.On this basis,the GILncSig was constructed and was used to assess the prognostic risk of HCC patients.The formula used was as follows: GILncSig=(0.0867 × expression level ofMIR210HG)+(0.0454 × expression level ofAC016735.1)+(0.1316 × expression level ofAC116351.1)+(0.3036×expression level ofAC010643.1)+(0.2557 × expression level ofLUCAT1).In the GILncSig,the coefficients of these 5 LncRNAs were all positive,and their high expression was associated with poor prognosis.This indicates that these LncRNAs are risk factors.
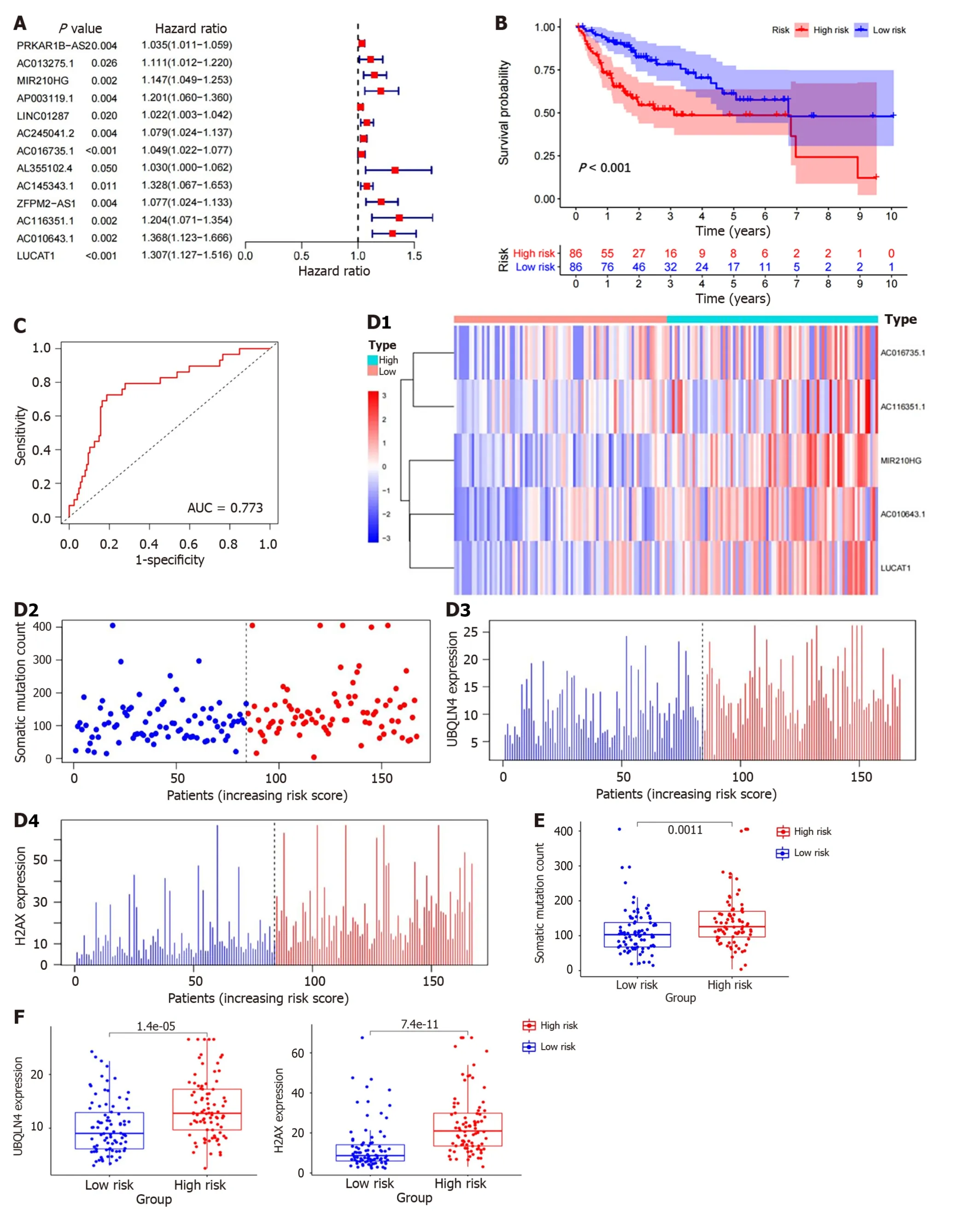
Figure 3 ldentification of the genomic instability-derived long non-coding RNAs signature in the training set. A: Forest plot: The P value,risk coefficient (HR) of 13 genomic instability (GI)-long non-coding RNAs (LncRNAs) in the training set analyzed by univariate Cox regression were significantly associated with hepatocellular carcinoma prognosis;B: Kaplan-Meier analysis of overall survival in patients with low or high risk according to the GI-derived LncRNAs signature (GILncSig) score in the training set;C: Time-dependent receiver operating characteristic curves analysis of the GILncSig;D: The LncRNA expression patterns,distribution of somatic mutations,UBQLN4 and H2AX expression with increasing GILncSig score;E: Somatic mutations count in the high-risk and low-risk groups for the training set patients.Red represents the high-risk group,and blue represents the low-risk group;F: The boxplots of UBQLN4 expression and H2AX expression between the high-risk and low-risk groups in the training group.
Risk scores were calculated for all patients in the training set using the GILncSig.Patients with risk scores equal to or higher than the median value were included in the high-risk group,and the remaining patients were included in the lowrisk group.Log-rank tests and K-M analysis showed that patients in the low-risk group had significantly better survival outcomes than those in the high-risk group (P< 0.001,Figure 3B).The 5-year survival rates in the two groups were 9.3% (high-risk group) and 19.8% (low-risk group).The ROC curve analysis of the GILncSig over time is shown in Figure 3C,and the AUC was 0.773.At the same time,GILncSig expression level,somatic mutation count and expression level ofH2AX,UBQLN4genes (a newly discovered driver of GI[23]) were also observed to change with an increase in the risk score (Figure 3D).For patients with high scores,the expression ofmiR210HG,AC016735.1,AC116351.1,AC010643.1andLUCAT1were up-regulated.Compared with the low-risk group,somatic mutations were more frequent in the high-risk group (P=0.0011,Figure 3E).In addition,the expression ofUBQLN4andH2AXwere higher in high-risk patients than in low-risk patients (P< 0.01,Figure 3F).
Independent validation of the GILncSig on the RNA-seq platform of HCC data
Subsequently,in order to examine the credibility of the prognostic performance of the GILncSig,we used the independent testing set of 171 patients to determine this.Similarly,using the GILncSig to calculate the risk score of patients in the testing set,the patients were also divided into the high-risk group (n=76) and low-risk group (n=95) according to the same method as in the training set,and the K-M analysis also showed significant differences between the two groups.The OS rate in the low-risk group was significantly better than that in the high-risk group (P=0.013,Figure 4A).The 5-year survival rate in the high-risk group was 3.95%,which was lower than that in the low-risk group (12.63%).In the testing set,ROC curve analysis of the GILncSig over time showed that the AUC was 0.679 (Figure 4B).Similar to the training set,the expression of GILncSig as well as somatic mutation count and the expression ofH2AX,UBQLN4in the testing set were mostly positively correlated with the risk value (P< 0.01,Figure 4C).The somatic cell mutation rate of the high-risk group in the testing set was slightly higher than that of the low-risk group (P=0.18,Figure 4D).The expression level ofUBQLN4andH2AXin the low-risk group was significantly lower than that in the high-risk group (Figure 4E,P< 0.01).
Similarly,we divided all patients in the TCGA set into the high-risk group (n=162) and low-risk group (n=181) and used the same method to verify the performance of the GILncSig.As expected,we obtained similar but more meaningful results.The OS rate and 5-year survival rate (6.79% to 16.02%) of patients in the high-risk group were lower than those in the low-risk group (P< 0.01,Figure 4F).ROC curve analysis of the GILncSig in the TCGA set over time showed that the AUC value was 0.730 (Figure 4G).Figure 4H shows the expression of GILncSig,somatic mutation count and the expression ofUBQLN4,H2AXin the TCGA set.As expected,the somatic cell mutation rate and the expression levels ofUBQLN4andH2AXin the high-risk group were significantly higher than those in the low-risk group (Figure 4I,P=0.0011;P< 0.01,respectively).
Comparison of the prediction ability of the GILncSig with existing LncRNA signatures
The predictive performance of the GILncSig in our study was then compared with two published LncRNA signatures for predicting HCC prognosis: 6-LncRNA signature derived from Gu’s study (hereinafter referred to as GuLncSig)[24] and 4-LncRNA signature derived from Wu’s study (hereinafter referred to as WuLncSig)[25] using the same TCGA patient cohort.On this basis,ROC curve analysis was used to evaluate the prognostic performance of these signatures.As shown in Figure 5,the AUC of the the GILncSig was 0.736,which was higher than that of GulncSig (AUC=0.664) and WulncSig (AUC=0.725).These results may indicate that the GILncSig has better prognostic performance than the two recently published LncRNA signatures.
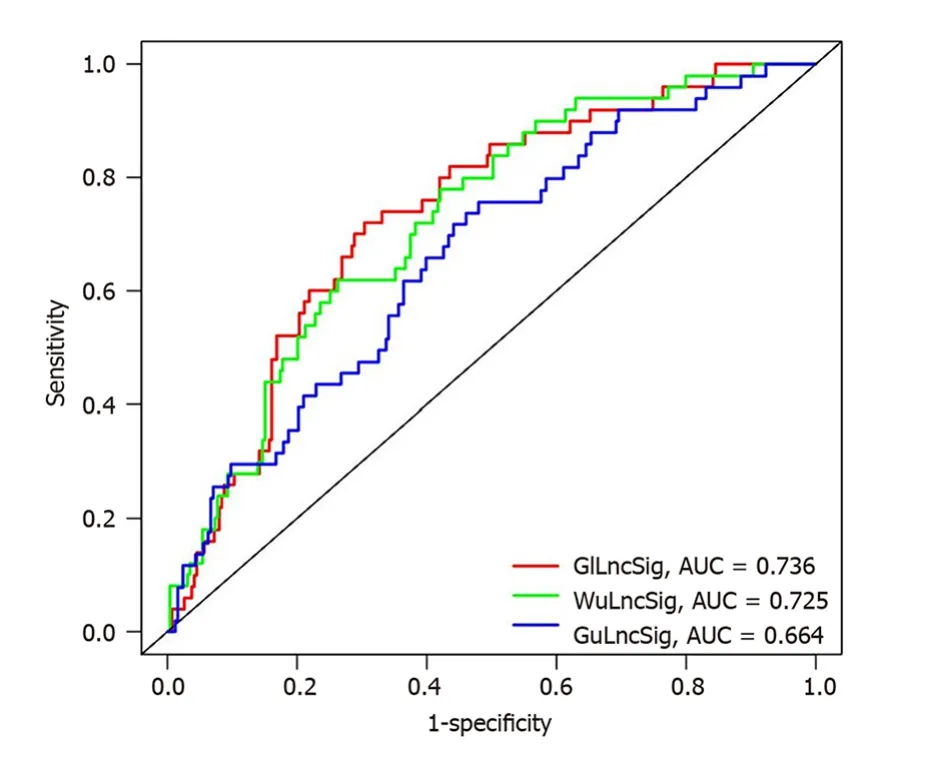
Figure 5 Receiver operating characteristic analysis was used to evaluate the performance of the genomic instability-derived long noncoding RNAs signature,genomically unstable-derived long non-coding RNAs signature,and WuLncSig. The area under the curve of overall survival for the genomic instability-derived long non-coding RNAs (LncRNAs) signature,genomically unstable-derived LncRNAs signature and WulncSig was 0.736,0.664 and 0.725,respectively.GILncSig: Genomic instability-derived long non-coding RNAs signature;GuLncSig: Genomically unstable-derived long non-coding RNAs signature;WuLncSig: LncRNA signature derived from Wu’s study;AUC: Area under the curve.
Independence of the GILncSig from other clinical factors
To verify whether the GILncSig can be used as an independent clinical variable to evaluate the prognosis of HCC patients,multivariate Cox regression analyses were performed for age,sex,grade,stage,and prognostic risk score based on the GILncSig.The GILncSig was found to be statistically significant as an independent prognostic factor (P< 0.05,Table 1).To determine whether the GILncSig can be applied to different clinical traits,we first divided the TCGA group into groups older than 65 years (n=141) and younger than or equal to 65 years (n=235) and the risk scores of patients in each age group were calculated by the GILncSig.Patients in each group were divided into high-risk and low-risk groups according to the median risk score.The results showed significant differences in survival between the two groups (P< 0.01,Figure 6A).Next,TCGA patients were also divided into the male group (n=255) and female group (n=122) and then the patients in each group were divided into the high-risk group and low-risk group by the GILncSig.In the male group,the difference in OS between the high and low risk groups was considered significant and meaningful,whereas in the female group,the result was not significant (Figure 6B,P< 0.001,P=0.952).We next used the same method to divide patients into two groups according to other clinical conditions,such as grade,stage,T stage,M stage,N stage,and then divided them into the high and low risk group using the GILncSig.As expected,Figure 6C-G shows that in most clinical subgroups,the OS of low-risk patients was significantly better than that of high-risk patients,including Grade 1-2 (P< 0.001),M0 (P< 0.001),N0 (P< 0.001),T1-2 (P=0.002),and Stage I-II (P=0.006).However,the results in the M1,N1-3 and stage III-IV were seemingly meaningless (P> 0.1),and thePvalue was only slightly significant in Grade 3-4 (0.089) and T3-4 (P=0.085).
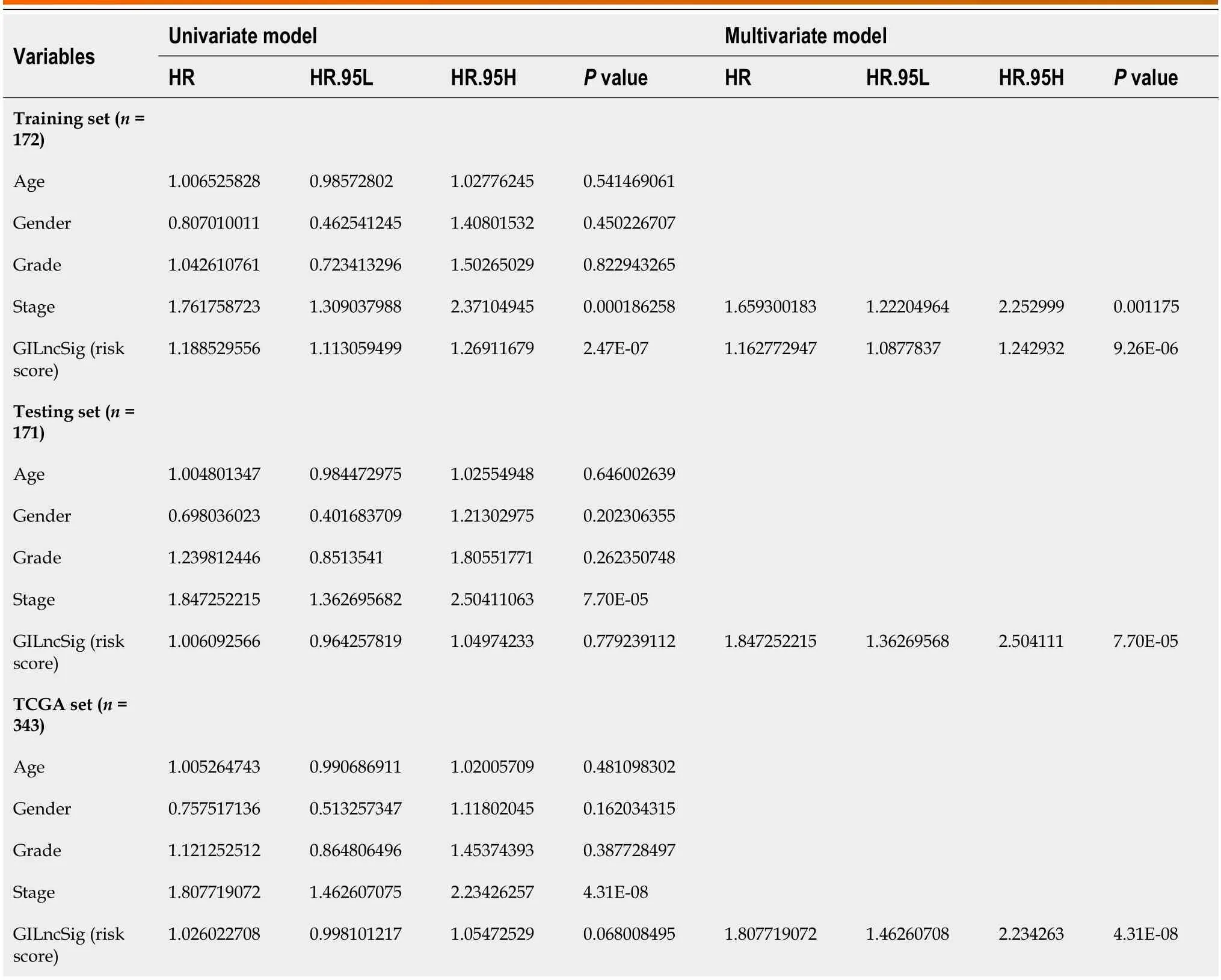
Table 1 Univariate and multivariate Cox regression analysis were performed for the risk score models which were based on the genomic instability-derived long non-coding RNAs signature and the overall survival of each patient group
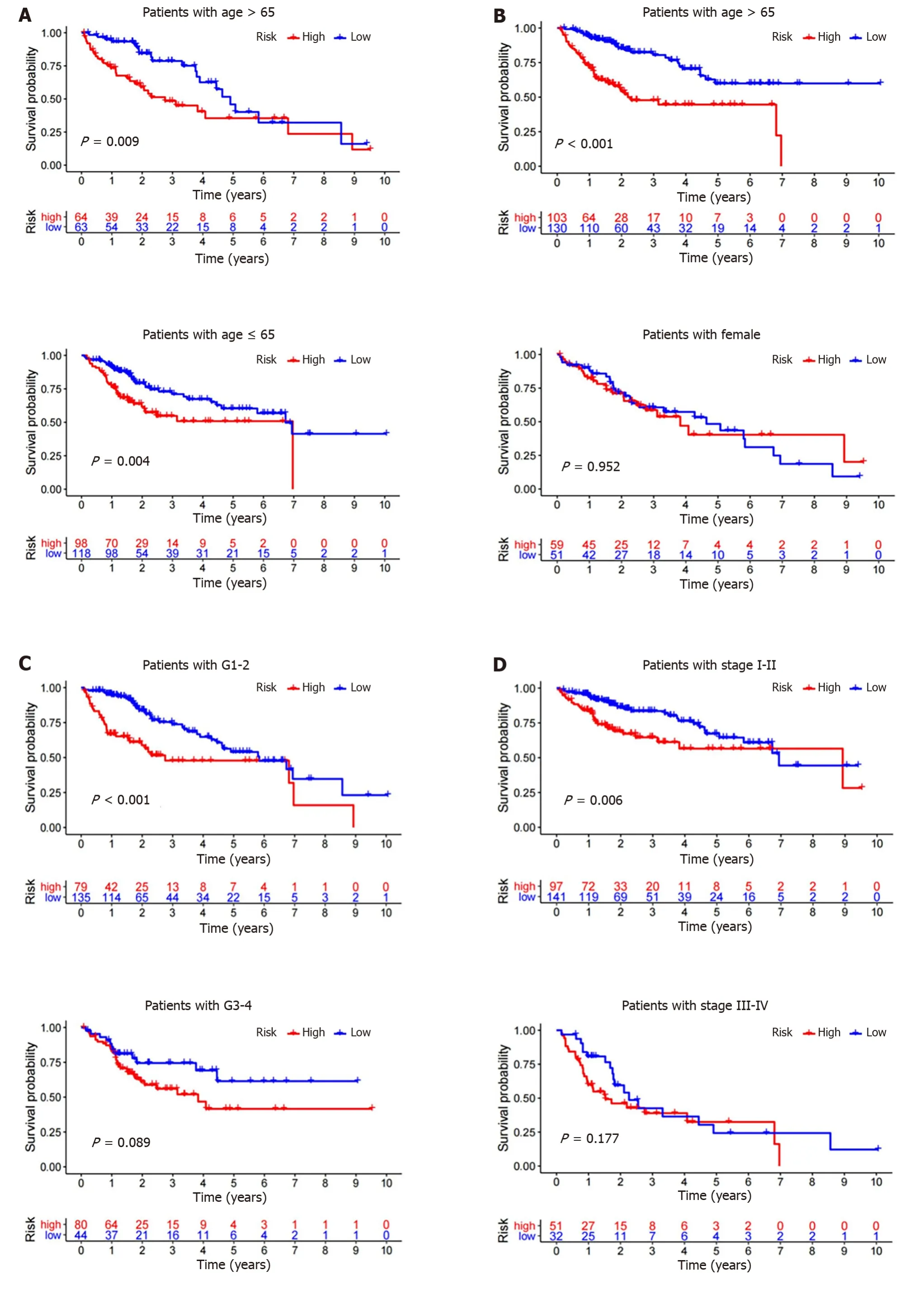
Figure 6 Kaplan-Meier survival analyses of patients with different clinical characteristics. Kaplan-Meier curve analysis of overall survival in the high-risk and low-risk groups.A: Age older than 65 years and age younger than or equal to 65 years;B: Male and female;C: Grade 1-2 and Grade 3-4;D: Stage I-II and stage III-IV;E: T1-2 and T3-4;F: M0 and M1;G: N0 and N1-3.
These findings may mean that the GILncSig can be used as a reliable independent prognostic factor to predict the prognosis of HCC patients.It appears to be a better predictor of prognosis for HCC patients in the early stages of the disease.
Further exploration of the predictive power of the GILncSig
TP53 mutation is the most common mutation in HCC,and it affects the progression and prognosis of the disease[26].Mutations in TP53 are closely related to poor survival in HCC patients,and can be used as an independent prognostic biomarker in HCC[27].As shown in Figure 7A,the percentage of patients with TP53 mutations was 51%,43% and 47% in the high-risk groups of the training set,testing set and TCGA set,respectively,which were significantly higher than 21%,12% and 16% in the low-risk group in each set.This suggests that the GILncSig is also associated with TP53 mutation status.In addition,K-M survival analysis of TCGA patients was further performed in combination with TP53 mutation status and the GILncSig.As expected,patients in the TP53 wild-type combined with GS-like group had the best prognosis and those in the TP53 mutant combined with GU-like group had the worst prognosis.Patients with the same TP53 mutation status had a better prognosis than those in the GU-like group (P=0.009,Figure 7B).These results suggest that the GILncSig may have a more reliable predictive power for HCC patients than TP53 mutation status alone.
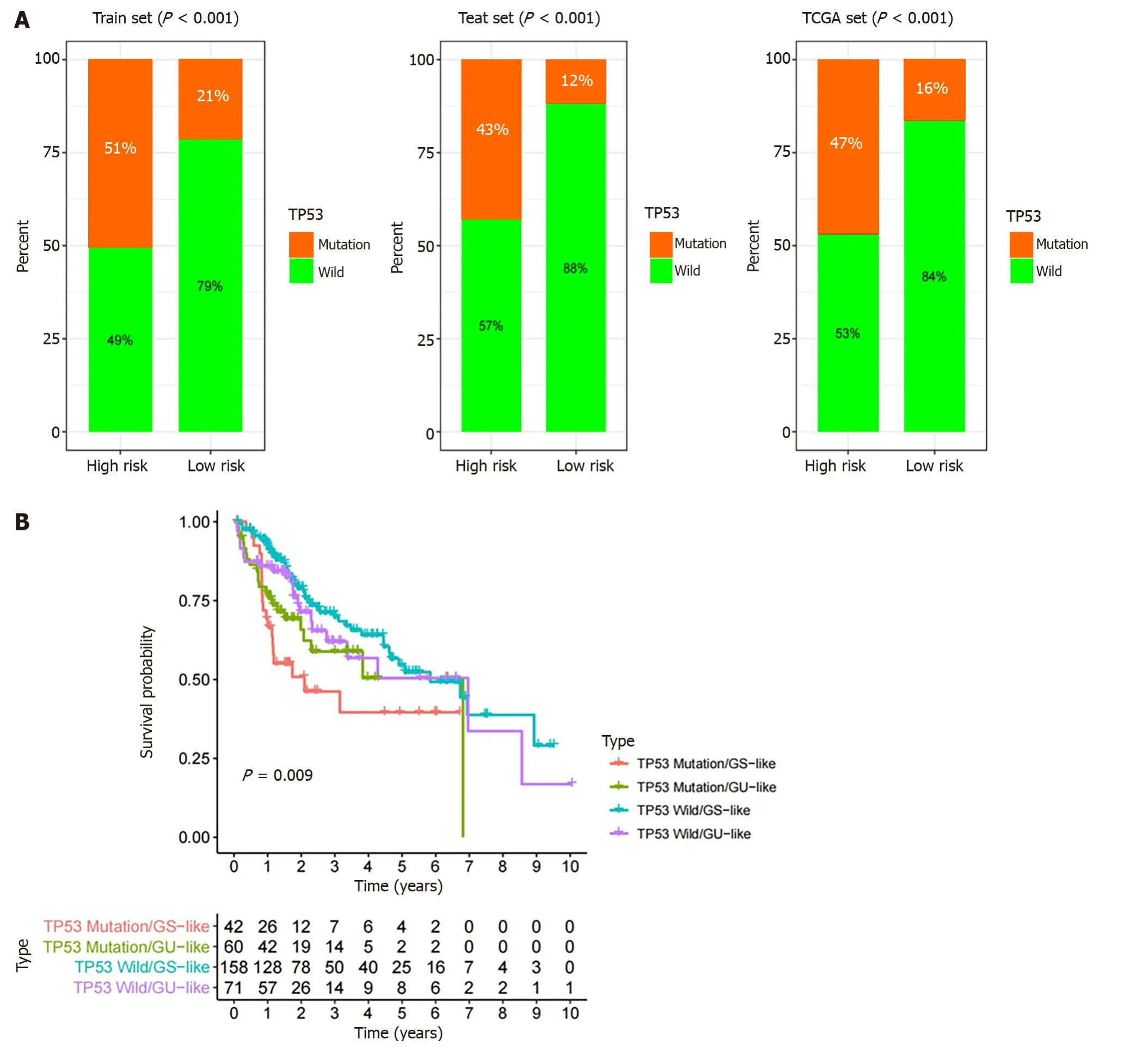
Figure 7 Comparison of the genomic instability-derived long non-coding RNA signature with TP53 mutation status for prognosis. A: The proportion of TP53 mutation in the high-and low-risk groups in the training set,testing set and the TCGA set;B: Kaplan-Meier curve analysis of overall survival based on TP53 mutation status and genomic instability-derived long non-coding RNA signature classification.GU: Genomically unstable;GS: Genomically stable.
DlSCUSSlON
In the past few years,a large number of studies have been conducted on the initiation,diagnosis and treatment of HCC[28,29].At present,traditional clinicopathological features are still used as a tool to predict the prognosis of HCC[30].An imaging examination is essential for the diagnosis of liver cancer,but the sensitivity of imaging will be greatly reduced due to the small lesions and insignificant symptoms of early liver cancer[31].In recent decades,among all biomarkers for the diagnosis of HCC,alpha-fetoprotein (AFP) is the most widely used and relatively reliable.Abnormal plasma AFP level is closely related to HCC malignancy[32].However,due to its lack of sensitivity and specificity,the results are unsatisfactory in the diagnosis of early liver cancer[33].Therefore,the identification of new reliable prognostic indicators is urgently required to evaluate the prognosis of HCC patients.
In recent years,with the rapid development of high-throughput sequencing technology,GI-LncRNAs have been gradually identified as potential prognostic indicators[16,17].It is reported that GI is one of the ubiquitous characteristics of cancer[10,34,35].It also has great potential as one of the prognostic factors in HCC patients[12].In addition,aberrant expression of LncRNAs may affect cell proliferation,tumor progression or metastasis,suggesting that LncRNAs may also be new prognostic factors for HCC by affecting GI[36].A considerable number of studies have found that some LncRNAs are associated with gene instability,thus affecting the prognosis of cancer,such as MANCR[37],CCAT2[38] and NORAD[17].Nevertheless,it is still difficult to identify GI-LncRNAs,their significance in predicting the clinical outcome of HCC is unclear,and their potential as a new prognostic marker remains to be explored.Thus,we constructed a computational framework for identifying genome instability-related LncRNAs by combining LncRNA expression with tumor mutant phenotype.
In this study,we first obtained 88 GI-LncRNAs by comprehensive analysis of the LncRNA profile and somatic mutation downloaded from TCGA database.PCGs closely associated with LncRNAs were identified and analyzed for functional enrichment.Through KEGG and GO pathway analysis,we found that their biological processes and biological pathways mainly involved the small molecule catabolic process and fatty acid metabolic process,pyrimidine metabolism,purine metabolism,and folate biosynthesis.Pyrimidine metabolism,purine metabolism and folate biosynthesis are involved in DNA synthesis.Dysfunction related to DNA damage will lead to cell cycle imbalance and GI[39].In addition,the Fanconi anemia pathway is composed of a complex DNA damage signal and repair network,which is very important in preventing GI[40].
In addition,we obtained five GI-LncRNAs (miR210HG,AC016735.1,AC116351.1,AC010643.1andLUCAT1),and further explored the roles these GI-LncRNAs play in predicting the clinical outcome of HCC patients.Based on this,the GILncSig was established.Subsequently,the GILncSig was used to divide the patients into two groups with high and low risk.In the training set,patients in the low-risk group survived longer than those in the high-risk group,and the independent TCGA set and testing set further validated this result.The area under the ROC curve of the GILncSig in the three groups mentioned above was 0.773,0.679 and 0.736 respectively,which demonstrated that the GILncSig has excellent prognostic ability.In all HCC cohorts,we found that the number of somatic mutations was higher in the highrisk group than in the low-risk group.In addition,the expression ofUBQLN4andH2AXwas significantly higher in highrisk patients than in low-risk patients.UBQLN4is an identified driver of gene instability in a variety of cancers,and its overexpression in HCC tissues leads to poor prognosis[23,41].A recent study indicated that HCC patients with high expression ofmiR210HGhad a worse prognosis than those with low expression[42].LUCAT1has also been found to be directly associated with the development and progression of cancers,including HCC,and its inhibition ofANXA2phosphorylation in HCC promotes tumorigenesis[43,44].AC010643.1andAC116351.1have been used as key components of the recently published LncRNA signatures for predicting HCC prognosis,suggesting that they have great potential as new prognostic markers[25,45,46].However,little is known aboutAC016735.1.In general,these 5 LncRNAs play a crucial role in the pathogenesis of cancer and have potential prognostic value.TP53 is a common mutation site in cancer,and its mutation type is significantly associated with lower survival rate of HCC patients[47,48].According to the GILncSig,the mutation rate of TP53 in high-risk patients was significantly higher than that in low-risk patients.In addition,there was a significant difference in survival between high-risk and low-risk patients with TP53 mutations.Therefore,it is of great significance for personalized prognostic evaluation of HCC patients.
Many previous studies have used similar methods to find prognosis-related LncRNAs and establish LncRNA signatures to predict the prognosis of HCC,such as the studies by Huanget al[49] and Wuet al[25].Moreover,as all data used in this study were collected from TCGA database,similar results could be obtained when searching for GI-LncRNAs and exploring their functional pathways.The difference is that all HCC patients were divided into the training set and the testing set according to the principle of random grouping.As a result,the calculated prognosis-related LncRNAs were different,and the established formula of the GILncSig was also different.In addition,the AUC of the GILncSig in this study was relatively high.Subsequently,the GILncSig showed good performance in both the independent testing set and TCGA set.Although this study quantified the GI index of HCC patients and established the GILncSig to assess patient outcomes,there are still some limitations that need to be further investigated.Firstly,the GILncSig was based on a single TCGA database,which requires an independent,large and comprehensive database for further verification.Due to the limited availability of LncRNAs in HCC samples in the GEO database,we did not use the GEO database for further study.In addition,the GILncSig was determined using the computational framework based on mutation hypothesis.In the future,in vivoorin vitroexperiments are needed to verify its mechanism in the development of liver cancer.
CONCLUSlON
We established a computational framework for identifying genome instability-related LncRNAs by combining LncRNA expression with tumor mutant phenotype,which can be used as an independent biomarker to predict the clinical outcome of HCC patients.This is helpful for prognosis assessment and further clinical decision-making in HCC patients.
ARTlCLE HlGHLlGHTS
Research background
Long stranded non coding RNA (LncRNA) has been found to be a potential prognostic factor in cancer,including hepatocellular carcinoma (HCC).Some LncRNAs have been confirmed as potential indicators for quantifying genomic instability (GI).However,GI-LncRNAs have yet to be largely explored.This study established the GI-derived LncRNA signature (GILncSig),which can predict the prognosis of HCC patients.
Research motivation
We established a GILncSig that can predict the prognosis of HCC patients,which can help to guide prognostic evaluation and treatment decisions.
Research objectives
The aim of this study was to establish a GILncSig for predicting the prognosis of HCC patients.At present,the treatment of liver cancer has achieved certain results.However,existing research evidence suggests that the treatment options currently used in clinical practice are still relatively ineffective.The objective effective rate of treatment is still largely inadequate,and most patients do not have good responses.The 5-year overall survival of metastatic HCC is still not ideal.Further research should mainly focus on expanding treatment targets and searching for reliable biomarkers,which will help adjust treatment choices and avoid the risks and costs associated with drug ineffectiveness and side effects.Therefore,there is an urgent need for new biomarkers to predict the prognosis of HCC patients.
Research methods
GI-LncRNAs were identified by combining LncRNA expression and somatic mutation profiles.Next,GI-LncRNAs were analyzed for functional enrichment.The GILncSig was established in the training set by Cox regression analysis,and its predictive ability was verified in the testing set and TCGA set.In addition,we explored the effects of the GILncSig and TP53 on prognosis.
Research results
A total of 88 GI-LncRNAs were found,and functional enrichment analysis showed that their functions were mainly involved in small molecule metabolism and GI.The GILncSig was constructed by 5 LncRNAs (miR210HG,AC016735.1,AC116351.1,AC010643.1,LUCAT1).In the training set,the prognosis of high-risk patients was significantly worse than that of low-risk patients,and similar results were verified in the testing set and TCGA set.Multivariate Cox regression analysis and stratified analysis confirmed that the GILncSig could be used as an independent prognostic factor.ROC curve analysis of the GILncSig showed that its area under the curve (0.773) was higher than the two LncRNA signatures published recently.Furthermore,the GILncSig may have a better predictive performance than TP53 mutation status alone.
Research conclusions
We established a GILncSig that can predict the prognosis of HCC patients,which will help to guide prognostic evaluation and treatment decisions.
Research perspectives
It is necessary to find new reliable biomarkers to predict the prognosis of HCC patients,adjust the treatment plan,and avoid the risks and costs associated with drug ineffectiveness and side effects.
FOOTNOTES
Co-first authors:Bo-Tao Duan and Xue-Kai Zhao.
Author contributions:Duan BT and Zhao XK drafted the manuscript;Zhou L and Zhang XY provided guiding advice on manuscript editing.All authors approved the final version of the manuscript.
lnstitutional review board statement:No human or animal research was included in this study.
Clinical trial registration statement:This study is a bioinformatics article and does not involve clinical trials.There are no relevant participants involved.
lnformed consent statement:No human research was included in this study.
Conflict-of-interest statement:The authors declare that they have no competing interests.
Data sharing statement:No additional data are available.
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers.It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BY-NC 4.0) license,which permits others to distribute,remix,adapt,build upon this work non-commercially,and license their derivative works on different terms,provided the original work is properly cited and the use is non-commercial.See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:China
ORClD number:Bo-Tao Duan 0000-0003-4892-0036;Xue-Kai Zhao 0000-0002-7079-9095;Lin Wang 0000-0002-1834-8078;Lei Zhou 0000-0003-4615-645X;XingYuan Zhang 0000-0002-1817-8048.
S-Editor:Fan JR
L-Editor:Webster JR
P-Editor:Yu HG
 World Journal of Gastrointestinal Surgery2024年3期
World Journal of Gastrointestinal Surgery2024年3期
- World Journal of Gastrointestinal Surgery的其它文章
- Alcohol associated liver disease and bariatric surgery: Current perspectives and future directions
- Applications of gastric peroral endoscopic myotomy in the treatment of upper gastrointestinal tract disease
- Ex vivo liver resection and auto-transplantation and special systemic therapy in perihilar cholangiocarcinoma treatment
- Evaluation of bacterial contamination and medium-term oncological outcomes of intracorporeal anastomosis for colon cancer: A propensity score matching analysis
- Rescue from complications after pancreaticoduodenectomies at a low-volume Caribbean center: Value of tailored peri-pancreatectomy protocols
- Comparison of prognosis and postoperative morbidities between standard pancreaticoduodenectomy and the TRlANGLE technique for resectable pancreatic ductal adenocarcinoma