
液相色谱—串联质谱法测定蜂蜜中4种喹诺酮类药物残留量的不确定度评定
2024-05-06 15:05杨韵
食品与机械 2024年3期
杨 韵
曹 阳1,2
李 菁1,2
钟菲菲1,2
左 贺3
(1. 长沙市食品药品检验所,湖南 长沙 410036;2. 湖南酒类产品质量检验检测中心〔湖南〕,湖南 长沙 410036;3. 湖南劳动人事职业学院,湖南 长沙 410100)
中国有着悠久的养蜂史,是全球蜂蜜生产、贸易大国,产量和出口量均居世界首位[1]。随着现代畜牧业向规模化、集约化发展,蜂蜜中检出兽药的情况时有发生。2002年欧盟以抗生素(氯霉素)残留超标为由,全面禁止中国蜂蜜进入欧盟市场,因此,抗生素残留控制已成为食品安全工作中的重要一环,其检测技术也是食品安全研究领域亟待解决的课题[2-3]。
喹诺酮类药物是一种人工合成的含4-喹诺酮基本结构的类抗菌药物,主要是将拓扑异构酶与细菌DNA螺旋酶作为靶点,抑制DNA回旋酶促进DNA复制,对细菌DNA造成不可逆损伤以发挥抗菌效果,对多种革兰氏阴性菌的杀菌效果明显[4-6]。喹诺酮类药物具有抗菌活性强、抗菌谱广泛、不良反应少、体内分布良好等多重优势,被广泛应用于人和动物的疾病治疗[7]。腐臭病、败血病等细菌性疾病会严重影响蜜蜂产蜜的产量和质量,为了蜜蜂的健康,蜂农会使用喹诺酮类药物为蜜蜂“治病”,若产生抗药性,则会加大用药剂量,从而造成蜂蜜的药物残留[8]。研究[9]表明,长期食用含喹诺酮类药物残留的蜂蜜会导致人体中枢神经系统出现问题,严重者甚至出现肌无力的状况,并且还有一定的致癌性。
不确定度是以测量结果本身为研究对象,由于随机影响和系统影响的存在而对测量结果不能肯定的程度,只要是测量数据,都不可避免地存在不确定度,是客观存在的影响检测结果准确的因素[10-12]。为保证检测结果的准确性,可以通过不确定度的评定来判定试验结果的质量,因此不确定度也是测量结果的一部分,同时是评价测量活动质量的重要指标,是衡量检测结果准确性和可靠性的重要参数[13-14]。研究拟依据GB 31657.2—2021对蜂蜜进行检测,并参照现行有效的相关标准及指南对蜂蜜中喹诺酮类药物残留的检测结果进行不确定度评定[15-17],分析检测过程中各测量不确定度分量对结果的影响,找出需要重点关注的环节,为实验室内部质量控制提供依据。
1 材料与方法
1.1 材料与试剂
蜂蜜:抽检样品;
水:GB/T 6682规定的一级水;
甲醇、乙腈:色谱纯,德国MERCK公司;
甲酸:色谱纯,上海阿拉丁生化科技股份有限公司;
恩诺沙星(CAS No:93106-60-6)、盐酸沙拉沙星(CAS No:91296-87-6)、氧氟沙星(CAS No:82419-36-1)、环丙沙星(CAS No:85721-33-1):北京曼哈格生物科技有限公司;
其他试剂均为国产分析纯。
1.2 仪器与设备
液相色谱串联三重四极杆质谱仪:Triple Quad 5500+型,美国AB SCIEX公司;
固相萃取柱:Oasis PRiME HLB型,200 mg/6 mL,美国Waters公司;
分析天平:XS205DU型,德国Mettler toledo公司;
高速冷冻离心机:X1R型,赛默飞世尔科技有限公司;
氮吹仪:N-EVAP45型,美国Organomation公司。
1.3 方法
1.3.1 标准曲线的制备 分别精密称取恩诺沙星0.010 08 g、盐酸沙拉沙星0.011 26 g、氧氟沙星0.010 13 g、环丙沙星0.010 35 g,用0.5 mL氨水溶解,并用甲醇定容至10 mL容量瓶,配制成质量浓度为1 mg/mL的标准储备液;用移液管准确量取0.1 mL各标准储备液于100 mL容量瓶中,用甲醇定容,配制成质量浓度为1 μg/mL的混合标准中间液;分别准确吸取0.10,0.20,0.50,1.00,2.00,5.00 mL混合标准中间液于100 mL容量瓶中,用20%乙腈—甲酸水溶液(20 mL乙腈中加入80 mL 0.1%甲酸溶液)定容,配制成质量浓度分别为1,2,5,10,20,50 ng/mL的标准系列工作液,各取1.00 mL,分别加入经提取、净化处理的空白试料中,溶解残余物,上清液过微孔尼龙滤膜后,上机测定。
1.3.2 样品前处理
(1) 提取:称取2.024 3 g蜂蜜样品于50 mL聚丙烯离心管中,加入磷酸盐缓冲液20 mL,涡旋1 min,振荡10 min,10 000 r/min离心5 min,取上清液备用。
(2) 净化:固相萃取柱分别用10 mL甲醇、10 mL水活化,将备用上清液过柱,用5 mL水淋洗,抽干后依次用4 mL 5%的甲酸—甲醇和4 mL乙酸乙酯洗脱,抽干后收集洗脱液,40 ℃氮气吹干。用1.00 mL 20%乙腈—甲酸水溶液溶解残余物,超声1 min后涡旋混匀,上清液过尼龙滤膜,待测。
1.3.3 液相色谱条件 色谱柱为Agilent SB-C18,2.1 mm×100 mm,1.8 μm;柱温35 ℃;进样量10 μL;流速0.3 mL/min;流动相A为0.1%甲酸水溶液,流动相B为甲醇;按表1进行梯度洗脱。
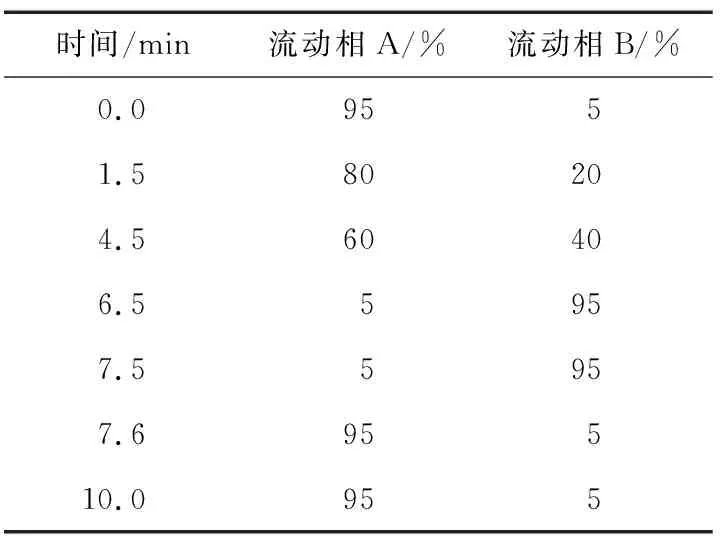
表1 梯度洗脱程序
1.3.4 质谱条件 电喷雾离子源;正离子扫描;多反应监测(MRM);电喷雾电压(IS)5 000 V;离子源温度(TEM)500 ℃;雾化气压力(GS1)379 kPa;雾化气压力(GS2)379 kPa;气帘气压力(CUR)207 kPa;碰撞器(CDA)62 kPa,质谱参数设置见表2。
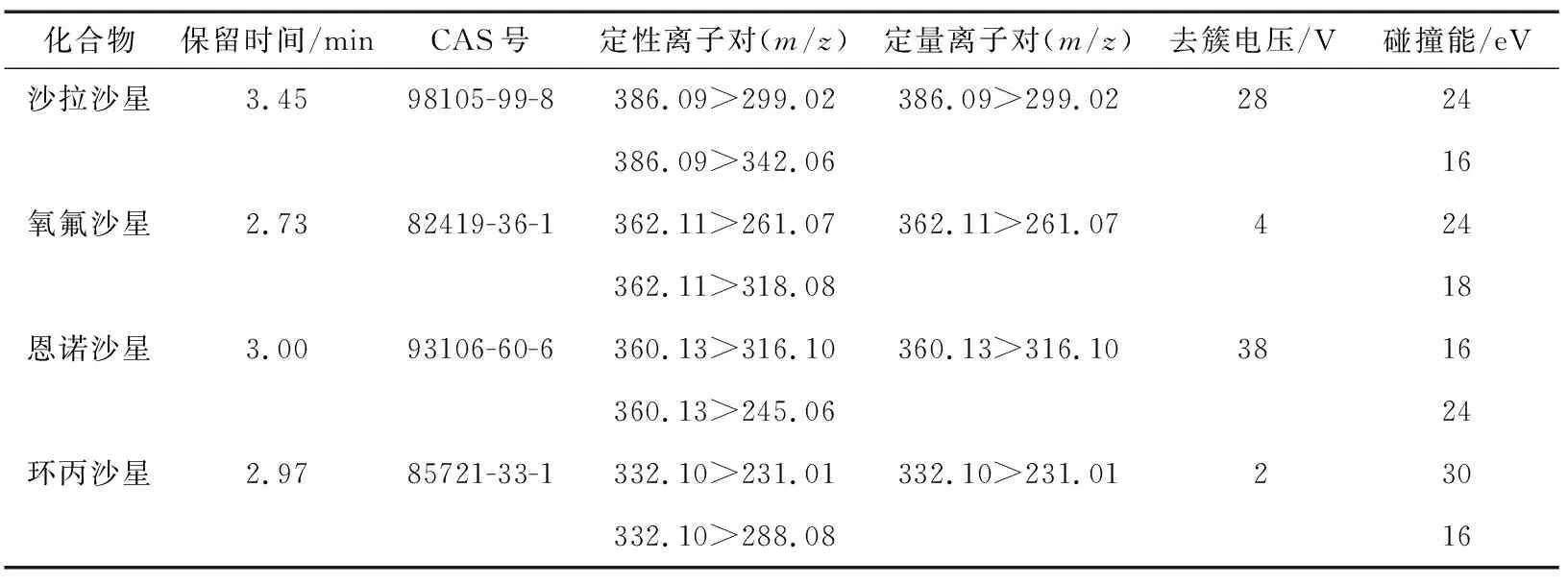
表2 多反应监测模式下6种化合物的质谱分析参数
1.3.5 数学模型建立 按式(1)建立数学模型。
(1)
式中:
X——样品中相应的喹诺酮类药物残留量,μg/kg;
Cs——基质标准溶液中相应的喹诺酮类药物质量浓度,ng/mL;
As——基质标准溶液中相应的喹诺酮类药物峰面积;
A——样品溶液中相应的喹诺酮类药物峰面积;
V——溶解残余物溶液体积,mL;
m——样品质量,g。
1.3.6 不确定度分量的主要来源分析 根据试验步骤和数学模型进行分析,影响样品中喹诺酮类药物残留量的测量不确定度主要有:标准溶液配制与稀释过程引入的不确定度;标准曲线拟合引入的不确定度;样品称样量引入的不确定度;样品定容体积引入的不确定度;测量重复性引入的不确定度;加标回收引入的不确定度。
2 结果与分析
2.1 标准溶液配制与稀释过程引入的不确定度urel(C)
2.1.1 标准物质纯度引入的相对标准不确定度urel(Cp)
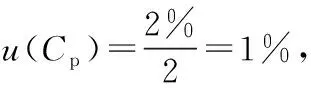
2.1.2 标准物质称量引入的相对标准不确定度urel(Cw)
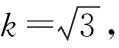
2.1.4 混合标准中间液制备过程引入的相对标准不确定度urel(Cs) 配制质量浓度为1 μg/mL的混合标准中间液需要用到1支1 mL A级分度吸量管,1个100 mL A级单标线容量瓶。取均匀分布,量具及温度波动引入的不确定度见表3。
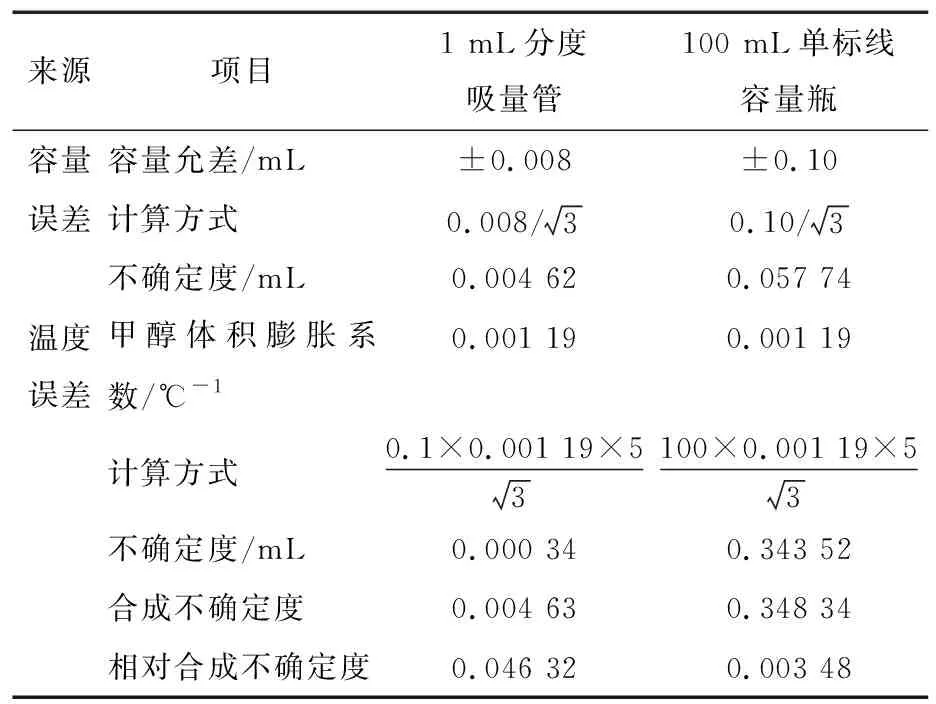
表3 混合标准中间液制备过程引入的不确定度
因此,由混合标准中间液制备过程引入的相对标准不确定度为:

(1)

表4 吸量管引入的不确定度
综上,标准系列工作液制备过程引入的相对标准不确定度为:
标准溶液配制与稀释过程引入的相对标准不确定度为:
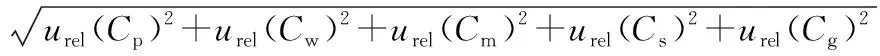
(2)
计算得:urel(C恩)=0.071 21;urel(C沙)=0.071 37;urel(C氧)=0.071 22;urel(C环)=0.071 22。
2.2 标准曲线拟合引入的不确定度urel(F)
标准系列工作液6个浓度,每个浓度测定3次,用最小二乘法拟合标准系列工作液各质量浓度与峰面积,以质量浓度为横坐标,峰面积为纵坐标,得到的线性回归方程见表5。被测样品重复测定6次,分别从标准曲线上拟合得到喹诺酮类药物残留量见表6。
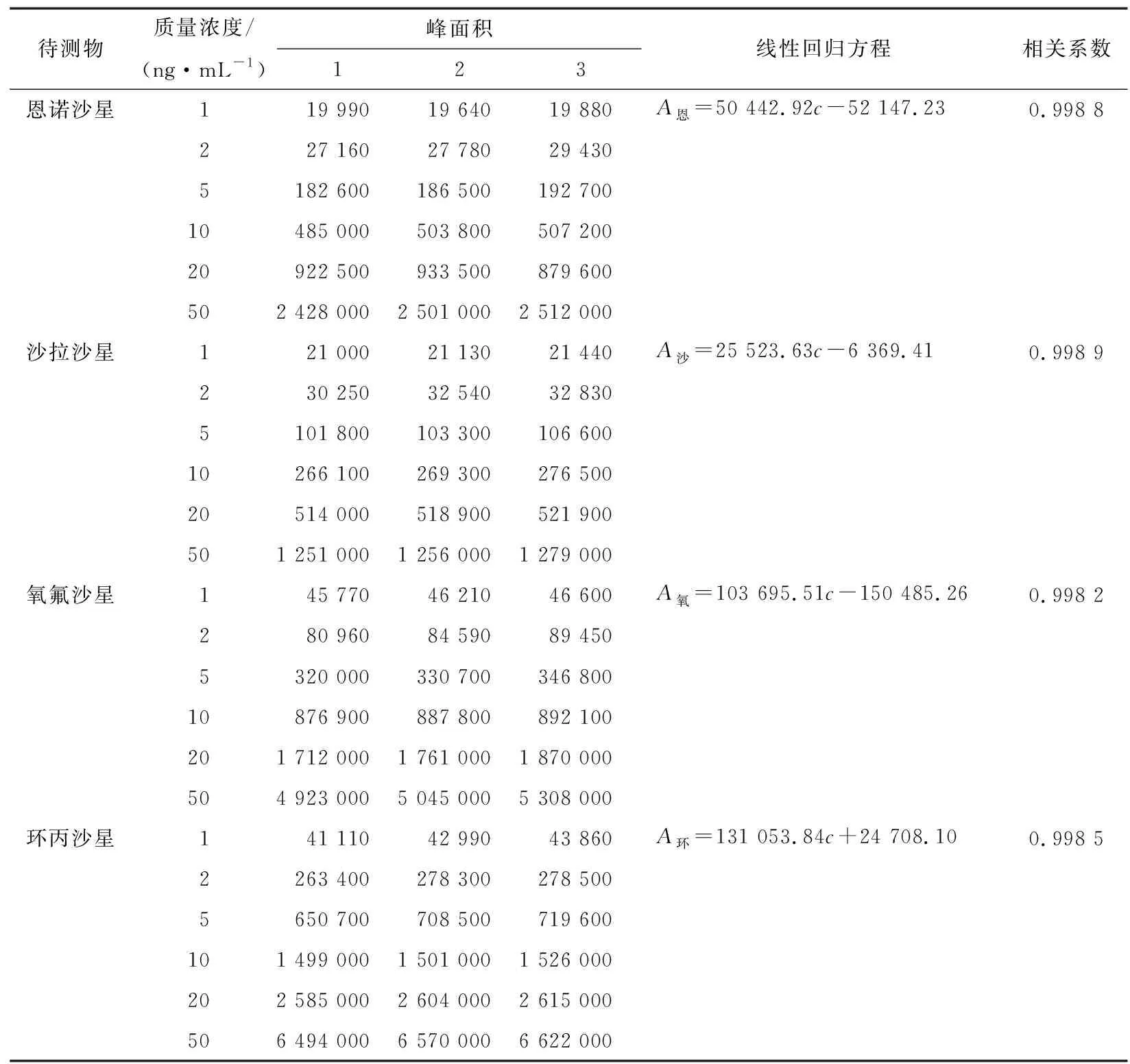
表5 标准曲线
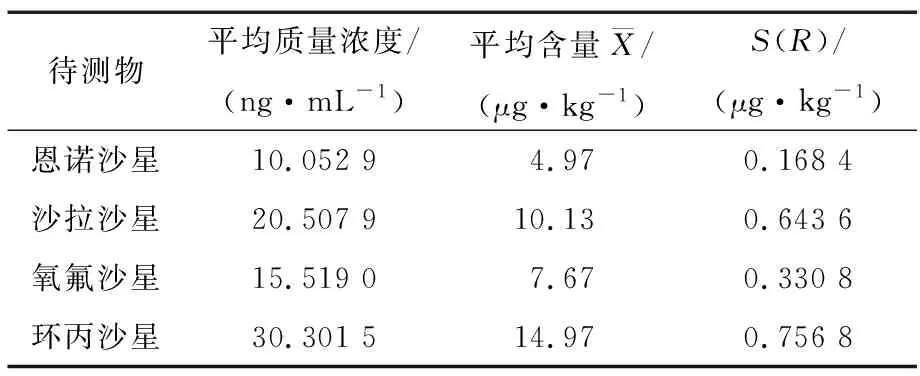
表6 样品重复测定数据
按式(3)、式(4)计算标准曲线拟合引入的相对标准不确定度。
(3)
(4)
式中:
urel(F)——标准曲线拟合引入的相对标准不确定度;
S——标准溶液峰面积比残差的标准差;
c0——由标准曲线拟合得到的样品中喹诺酮类药物残留量的平均质量浓度,ng/mL;
a——线性回归方程的斜率;
P——样品重复测定次数,P=6;
m——标准曲线的浓度个数,m=6;
n——标准曲线每个浓度溶液测定次数,n=3;

ci——第i个由标准曲线拟合的标准溶液质量浓度,ng/mL;
Aj——第j个标准溶液的峰面积;
cj——第j个标准溶液的配制质量浓度,ng/mL;
b——线性回归方程的截距。
将各值代入式(4)得:S恩=37 163.680 38,S沙=16 132.613 53,S氧=110 103.370 46,S环=96 300.170 38;urel(F恩)=0.034 86,urel(F沙)=0.014 74,urel(F氧)=0.032 26,urel(F环)=0.012 58。
2.3 样品称样量引入的不确定度urel(M)

2.4 样品定容体积引入的不确定度urel(V)

2.5 测量重复性引入的不确定度urel(R)
在空白蜂蜜样品中分别添加质量浓度为5,10,8,15 μg/kg的恩诺沙星、沙拉沙星、氧氟沙星和环丙沙星,从样品称量开始重复测定6次(n=6),测量结果见表6。用贝塞尔公式(5)~式(7)计算测量重复性引入的相对标准不确定度。
(5)
(6)
(7)
计算得:urel(R恩)=0.012 38,urel(R沙)=0.023 12,urel(R氧)=0.015 76,urel(R环)=0.018 39。
2.6 加标回收引入的不确定度urel(A)
取6份空白样品,在恩诺沙星5 μg/kg、沙拉沙星10 μg/kg、氧氟沙星8 μg/kg、环丙沙星15 μg/kg质量浓度下进行加标回收,样品回收率见表7。
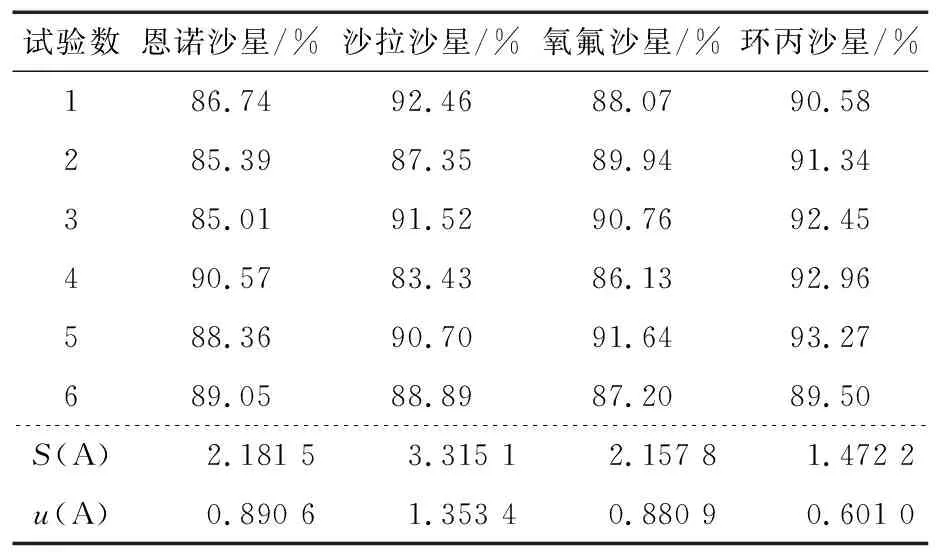
表7 样品回收率
计算得:urel(A恩)=0.010 18,urel(A沙)=0.015 20,urel(A氧)=0.009 90,urel(A环)=0.006 56。
3 合成标准不确定度与扩展不确定度
根据各不确定度分量得,相对合成标准不确定度为:
urel沙=0.078 09;urel氧=0.080 50;urel环=0.075 05。
根据RB/T 030—2020,以恩诺沙星为例,当恩诺沙星含量为4.97 μg/kg时,其合成标准不确定度u恩=4.97×0.081 02=0.403 μg/kg,扩展不确定度U恩=u恩×k=0.403×2=0.81 μg/kg。则u沙=0.791 μg/kg,U沙=1.58 μg/kg;u氧=0.617 μg/kg,U氧=1.23 μg/kg;u环=1.123 μg/kg,U环=2.25 μg/kg(取包含因子k=2,置信水平P=95%)。
4 测量结果及其不确定度报告
采用液相色谱—串联质谱法测定蜂蜜中喹诺酮类药物残留量,恩诺沙星、沙拉沙星、氧氟沙星、环丙沙星的测量不确定度结果分别表示为:X恩=(4.97±0.81) μg/kg;X沙=(10.13±1.58) μg/kg;X氧=(7.67±1.23) μg/kg;X环=(14.97±2.25) μg/kg。
5 结论
研究采用液相色谱—串联质谱法对蜂蜜中喹诺酮类药物残留量进行了测定,通过建立数学模型对试验过程中引入的不确定度各分量进行了评定。结果表明,标准溶液配制与稀释过程、标准曲线拟合对试验的不确定度结果贡献最大且不容忽视,其次是样品重复性测定和加标回收。因此,在测定蜂蜜中喹诺酮药物残留量的实际工作中,要确保标准溶液配制及稀释精准,选择精密度高的移液器和容量瓶,增加标准溶液、样品的测量次数,多做平行测定,提高检验操作技术,定期维护保养测量仪器,从而降低操作过程中各不确定度分量对检测结果的影响,提高检测结果的准确度。
方法GB 31657.2—2021包含了14种喹诺酮药物,试验只选取了其中4种较常见、易检出、具有一定代表性的药物,其中,2021年国家食品安全监督抽查计划将氧氟沙星列为蜂产品的风险监测项目。但该研究未考虑方法中其余10种药物的不确定度,加标浓度相对随机,且未考虑检测仪器对试验结果带来的影响,对实际工作的指导仍然存在一定的局限性。因此,在日常检测工作中应该以检出结果为目标,综合全面考虑不确定度来源,才能使检测结果更加准确、科学、规范。
猜你喜欢
世界最新医学信息文摘(2021年12期)2021-06-09
化工设计通讯(2020年10期)2020-09-17
大东方(2017年10期)2017-05-30
Cancer Biology & Medicine(2016年4期)2017-01-13
中国氯碱(2016年9期)2016-11-16
国外医药(抗生素分册)(2016年5期)2016-07-12
国外医药(抗生素分册)(2016年5期)2016-07-12
国外医药(抗生素分册)(2016年2期)2016-07-12
兽医导刊(2016年12期)2016-05-17
中国卫生标准管理(2015年25期)2016-01-14
