
凡纳滨对虾不同养殖模式下微生物群落结构分析
2024-04-30 18:14:51张芹吴小军杨兴丽
江苏农业科学 2024年6期
关键词:高通量测序
张芹 吴小军 杨兴丽
摘要:为了解不同养殖模式对凡纳滨对虾养殖环境微生物多样性和群落结构的影响,利用高通量测序技术,结合生物信息学分析,比较凡纳滨对虾主养模式和鱼-虾混养模式中水体、底泥以及虾肠道微生物的多样性和群落结构。结果显示,无论是物种的多样性水平还是丰富性水平,均是底泥>水>虾肠道,主养池塘底泥和虾肠道样品的物种多样性和丰富性均高于混养池塘。不同样品微生物群落的菌群分布于59个门,其中10个门的丰度>1%,变形菌门(Proteobacteria)和拟杆菌门(Bacteroidetes)属于绝对优势菌群,在混养池塘和主养池塘中均>10%。主养池塘的微生物标志物共有8个类群,分别为双歧杆菌目(Bifidobacteriales)、双歧杆菌科(Bifidobacteriaceae)、双歧杆菌属(Bifidobacterium)等,混养池塘的微生物标志物共有18个类群,分别为蓝色芽殖杆菌属(Gemmobacter)、外硫红螺菌科(Ectothiorhodospiraceae)、外硫红螺菌属(Ectothiorhodospira)等。通过功能预测及其相对丰度比较,发现主养模式在这些代谢通路中基因注释的相对丰度值均高于混养模式。以上研究表明,在凡纳滨对虾主养模式中添加的益生菌,有效提高了水体、虾肠道和底泥的微生物多样性,塑造了更健康、多元化、功能更强大的微生物群落,提高了养殖系统的物质循环能力和能量利用效率,更有利于虾体的健康成长。
关键词:凡纳滨对虾;微生物群落结构;高通量测序
中图分类号:S182;S917.1 文献标志码:A
文章编号:1002-1302(2024)06-0225-10
收稿日期:2023-05-02
基金项目:国家现代農业产业技术体系建设专项(编号:CARS-45);河南省农业科学院自主创新项目(编号:2023ZC113);国家淡水水产种质资源库建设项目(编号:FGRC:18537);河南省农业产业技术体系(编号:HARS-22-16-S)。
作者简介:张芹(1980—),女,河南辉县人,硕士,高级水产师,主要从事鱼类遗传育种研究。E-mail:zhangqin.2007@163.com。
凡纳滨对虾(Litopenaeus vannamei)别称南美白对虾,俗称白对虾或白肢虾,原产于南美洲太平洋沿岸,主要分布于秘鲁北部至墨西哥湾沿岸海域,是世界上养殖产量最高的三大优良虾类之一。凡纳滨对虾具有生长速度快、抗病力强、耐粗饲及便于运输等优点,是集约化高产养殖的优良品种。近年来,凡纳滨对虾的淡水养殖发展较快,而在淡水养殖过程中,水质的调控显得尤为重要,如何保持养殖水体中良好的微生态环境是对虾能否健康成长的关键因素。养殖池塘由水体、鱼(虾)、底泥组成,水体是鱼(虾)赖以生存的环境,水环境的稳定性与水体的藻类和微生物组成密切相关,当水体微生物多样性下降时,就会增加鱼虾病害发生的风险[1]。底泥中有水体长期的沉积物,包含动植物残骸、食物残渣、动物粪便等各种有机质,底泥复杂的组成为各种细菌提供了生长环境[2]。
凡纳滨对虾的健康生长和养殖环境整体的微生物组成有着密切的关系,调节好水体生态环境就是保持良好稳定的微生物群落组成。利用高通量测序技术,陈玲等探究了稻虾共作对稻田水体微生物多样性和群落结构的影响[3];杜世聪等研究了凡纳滨对虾病害发生前后池塘浮游细菌群落多样性、组成及关键类群的动态变化[4];金若晨等研究了凡纳滨对虾养殖过程中肠道和养殖环境微生物群落的结构及变化[5]。16S rDNA高通量测序技术是开展微生物菌落分析的有效手段,该技术在鳗鲡(Anguilla japonica)、斑点叉尾(Ictalurus punctatus)、草鱼(Ctenopharyngodon idella)、暗纹东方鲀(Takifugu obscurus)、黄颡鱼(Pelteobagrus fulvidraco)和红鳍东方鲀(Takifugu rubripes)等大量水生生物微生态环境的研究中得到了广泛的应用[6-11]。
本研究采取高通量测序技术,对2种不同模式凡纳滨对虾的养殖水体、底泥和虾肠道的微生物群落进行研究,以期为淡水养殖凡纳滨对虾的水质调控提供思路,为多元化的凡纳滨养殖模式提供技术支持。
1 材料与方法
1.1 试验池塘情况
试验地点在商丘梁园区文峰水产养殖发民专业合作社基地,凡纳滨对虾主养模式(zy):基地西区1号池塘,面积0.267 hm2,池塘消毒5 d后全池泼洒乳酸菌和小球藻(乳酸菌22.5 L/hm2+小球藻 7.4 L/hm2),同时泼洒复合维生素制剂,消毒1周后于7月2日投放淡化后的凡纳滨对虾虾苗12万尾。定期改底,每5~7 d使用10%~15%多硫酸氢钾复合盐全池泼洒,并在12 h后泼洒乳酸菌或EM菌。经过3个月养殖,10月初抽检凡纳滨对虾,平均体重约20 g,凡纳滨对虾体质健壮、颜色鲜亮。
鱼-虾混养模式(hy):基地东区7号池塘,面积1.33 hm2,主养草鱼(放养150 g以上大规格草鱼种1 000尾/667 m2),混养福瑞鲤2号夏花(2 000尾/667 m2)和凡纳滨对虾虾苗(4 000尾/667 m2),于2021年7月2日放养淡化好的虾苗,经过3个月养殖,10月11日抽检凡纳滨对虾,平均体重约 25 g,虾体质健壮,颜色鲜亮。
1.2 样品采集和检测
在2个池塘放入虾苗3个月后,虾基本养成,出池前进行抽检时分别采集水样(sy)、底泥(dn)和虾肠道样品(xcd)。采集饲料台附近距水体表层 10 cm 以下的水样,低温保存带回实验室,利用真空泵富集水体微生物,所用滤膜为孔径0.22 μm的醋酸纤维滤膜,抽滤体积为500 mL,抽滤完成后,滤膜-20 ℃保存。采用柱式采泥器采集池塘底泥,低温运回实验室后,-20 ℃保存,用于微生物分析。提前1 d采用地笼捕虾,采样时选择大小规格相近、健康无病伤、活动力强的个体进行样品采集,每个池塘取5尾凡纳滨对虾,解剖取肠道样品,混合后作为1个样本,-20 ℃保存。
1.3 样品DNA提取
底泥和肠道样品需要进行前处理:在研磨管中加入0.25 g样品,500 μL缓冲液Solution A,100 μL缓冲液Solution C和0.25 g研磨珠(肠道样品需要添加10 μL RNA酶),组织研磨仪上25 Hz研磨 10 min,水浴锅70 ℃裂解15 min,其间混匀1次,12 000 r/min 离心 2 min,转移上清液500 μL至新的离心管。
样品经过处理后,采用天根磁珠法土壤粪便DNA提取试剂盒和磁珠法天根通用型基因组DNA提取试剂盒分别从底泥、肠道和水样中提取基因组DNA。
1.4 PCR扩增和高通量测序
取30 ng基因组DNA样品,使用特异性引物进行PCR扩增,16S rDNA的V3+V4区引物序列为341F:5′-ACTCCTACGGGAGGCAGCAG-3′,806R:5′-GGACTACHVGGGTWTCTAAT-3′。扩增体系(25.0 μL):2×Taq PCR Master Mix 12.5 μL,Mix primer 2.0 μL,基因组DNA模板 1.0 μL,ddH2O 9.5 μL。扩增条件:98 ℃ 预变性 3 min;98 ℃ 变性10 s,55~70 ℃ 退火 20 s,72 ℃ 延伸30 s,15个循环;98 ℃ 预变性10 s,55 ℃ 退火20 s,72 ℃ 延伸 30 s,20个循环;72 ℃ 终延伸5 min,16 ℃ 保存。
对PCR产物进行2%琼脂糖电泳,使用磁珠对扩增产物进行纯化分选,采用Qubit 4.0荧光计进行定量,按固定组合将纯化的扩增产物进行等量混合,完成建库。采用生物分析仪对文库的片段范围及浓度进行检测,检测合格的文库在Miseq平台上采用PE250模式测序。
1.5 数据处理
测序获得raw reads后,进行组装过滤后得到effective tags,采用 Uparse 軟件对所有样品的全部effective tags 序列聚类,按照97%的一致性(identity)将序列聚类成为OTUs(operational taxonomic units),计算出每个OTU在各个样品中的Tags绝对丰度和相对信息,进行物种注释。
使用QIIME软件对样品进行α多样性分析,使用R语言工具绘制稀释型曲线、Venn图。运用Weighted Unifrac指数进行后续 β 多样性分析,利用R语言的Pheatmap包绘制物种分类热图,以热图形式展示组间weighted指数,利用Mothur软件将样本进行UPGMA分类树分类。根据OTU的物种注释和丰度信息,使用PICRUST软件进行KEGG Pathway的功能注释,并统计每个Pathway的丰度信息。
2 结果与分析
2.1 基因测序结果和α多样性指数
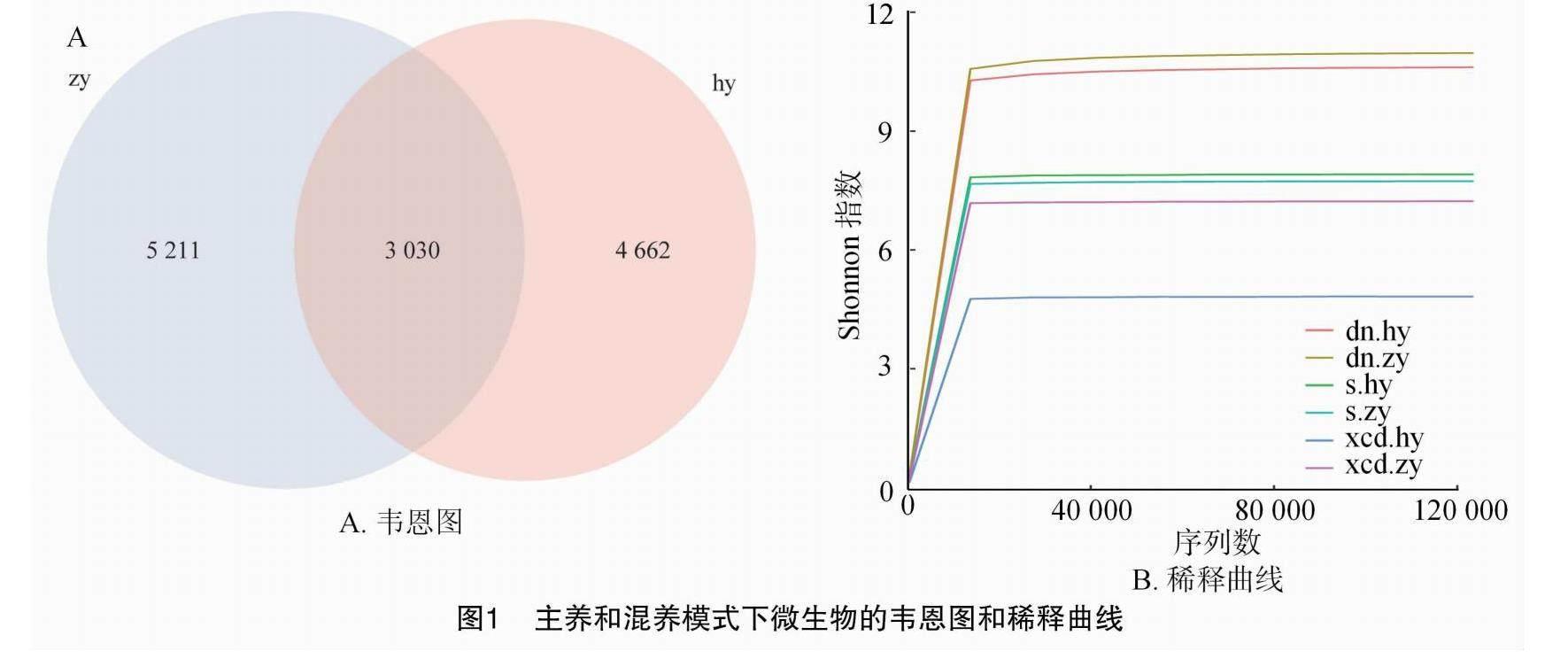
2种模式微生物测序结果显示,有效tags数量为1 223 411 条,最终得到的OTU数量为23 501 条,不同样本的OTU数量从1 146~8 889不等,2个池塘样品OTU数量均是底泥>水样>虾肠道。由图 1-A 可知,选取平均丰度>1的所有OTU进行韦恩图分析,其中,主养模式微生物包含5 211个独有OTU,混养模式微生物包含4 662 个独有OTU,在2种模式间共享的微生物共有3 030 个OTU。通过抽样n个tags来计算Shannon指数的期望值,然后根据一组n值与其相对应的Shannon期望值做出各个样品对应的Shannon稀释曲线,由图1-B可知,曲线趋于平缓,表明测序深度增加已不影响物种多样性,测序量趋于饱和,足够反映水体中的微生物信息。
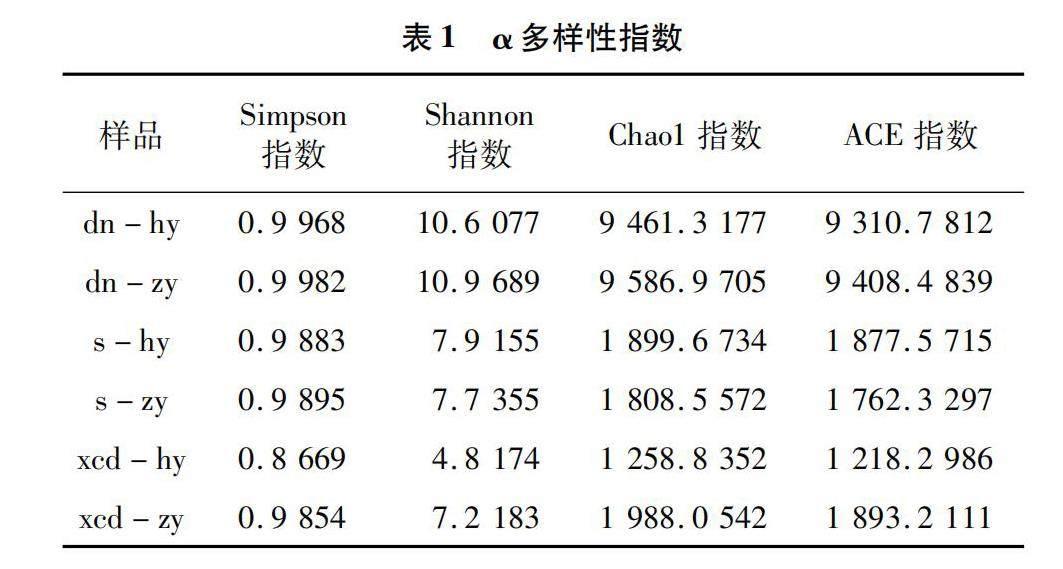
微生物的丰富度和均匀度可由α多样性指数反应,各指数的数值越大,代表物种的丰富度和均匀度越高。由表1可知,不同样品的Simpson指数在0.866 9~0.998 2之间,Shannon指数在4.817 2~10.968 9之间,Chao1指数在1 258.835 2~9 586.970 5 之间,ACE指数在1 218.298 6~9 408.483 9 之间。无论是Simpson指数还是Shannon指数均显示,物种的多样性水平顺序为底泥>水>虾肠道,其中,底泥样品的Simpson指数和Shannon指数均是主养塘>混养塘,水样的Simpson指数为主养塘>混养塘,而Shannon指数为混养塘>主养塘,虾肠道样品的Simpson指数和Shannon指数均是主养塘>混养塘,样品之间无显著性差异。Chao1指数和ACE指数均显示,混养池塘的物种丰富性为底泥>水>虾肠道,而主养池塘的物种丰富性为底泥>虾肠道>水;底泥的Chao1指数和ACE指数是主养塘>混养塘,水样的Chao1指数和ACE指数都是混养塘>主养塘,虾肠道的Chao1指数和ACE指数均是主养塘>混养塘,样品间无显著性差异。
2.2 基于门水平的细菌群落结构
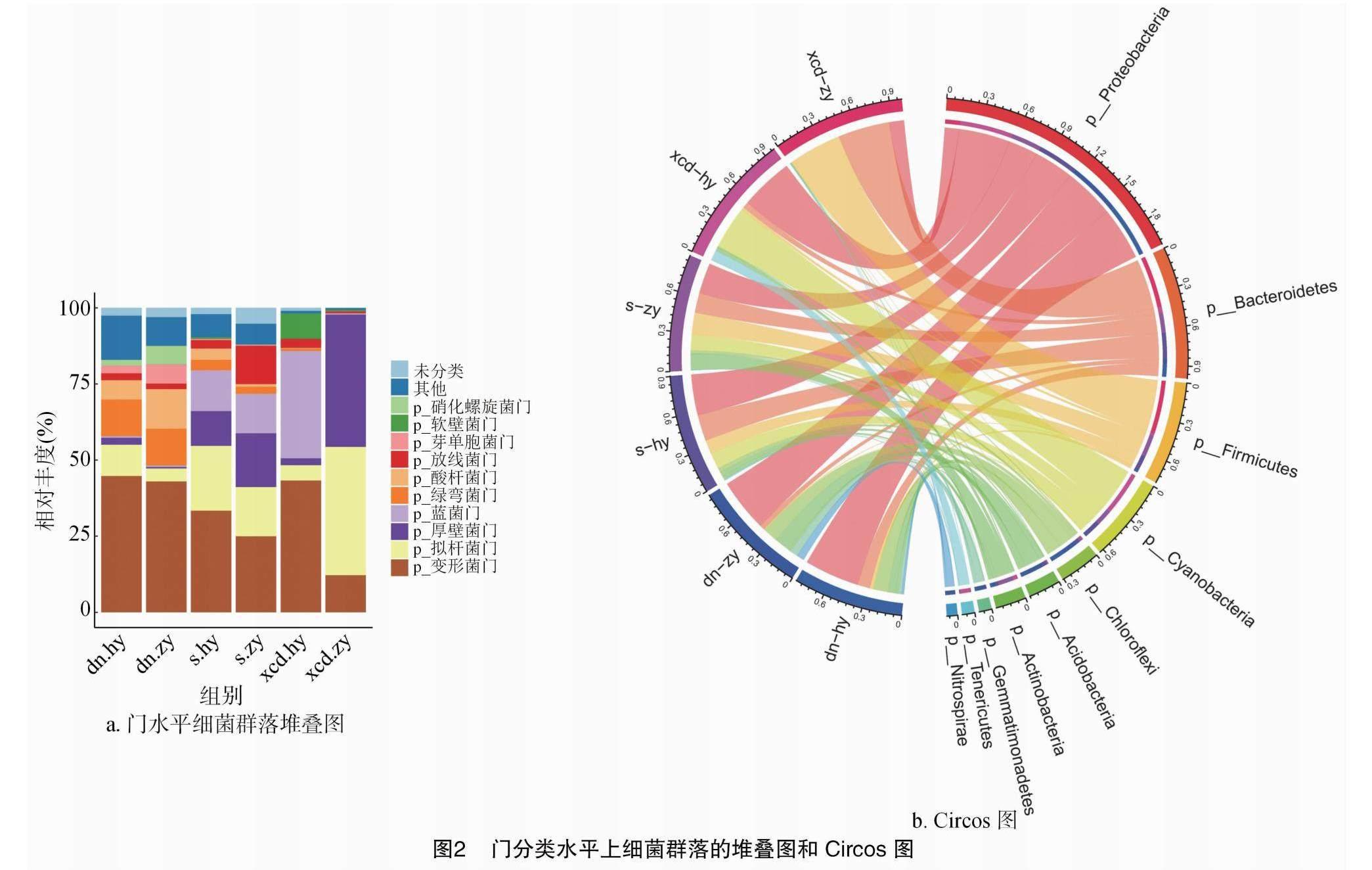
不同样品微生物群落菌群分布于59个门,其中,7个门的丰度>10%,10个门的丰度>1%。由图2可知,对丰度高于10%的门进行统计分析,其中,变形菌门(Proteobacteria)和拟杆菌门(Bacteroidetes)属于绝对优势菌群,在混养池塘和主养池塘中均>10%。
混养池塘底泥中优势菌群为变形菌门(44.7%)、绿弯菌门(Chloroflexi)(12.1%)、拟杆菌门(10.2%)和酸杆菌门(Acidobacteria)(6.3%),主养池塘底泥中优势菌群为变形菌门(43.0%)、酸杆菌门(13.0%)、绿弯菌门(12.1%)、芽单胞菌门(Gemmatimonadetes)(6.3%)、硝化螺旋菌门(Nitrospirae)(6.1%)和拟杆菌门(4.2%);混养池塘水体中的优势菌群为变形菌门(33.3%)、拟杆菌门(21.3%)、蓝菌门(Cyanobacteria)(13.4%)和厚壁菌门(Firmicutes)(11.4%),主养池塘水体中的优势菌群为变形菌门(24.9%)、厚壁菌门(17.6%)、拟杆菌门(16.2%)、蓝菌门(13.0%)和放线菌门(Actinobacteria)(12.6%);混养池塘虾肠道的优势菌群为变形菌门(43.3%)、蓝菌门(35.3%)、软壁菌门(Tenericutes)(8.4%)和拟杆菌门(5.0%),主养池塘虾肠道的优势菌群为厚壁菌门(43.4%)、拟杆菌门(42.2%)和变形菌门(12.1%)。
2.3 基于属水平的群落结构
不同样品微生物群落的菌群分布于683个属,其中,8个属的丰度>1%。由图3可知,混养池塘底泥中的优势菌群为硫杆菌属(Thiobacillus)(7.8%),水中的优势菌群为12up(5.4%)、Ruminococcaceae_NK4A214_group(1.9%)、志贺氏埃希菌属(Escherichia-Shigella)(1.4%)和Lachnospiraceae_NK4A136_group(0.9%),虾肠道中的优势菌群为气单胞菌属(Aeromonas)(9.6%)、盐单胞菌属(Cobetia)(7.5%)、弧菌属(Vibrio)(6.2%)和志贺氏埃希菌属(2.1%);主养池塘底泥中的优势菌群为硫杆菌属(3.2%),水中的优势菌群为hgcI_clade(4.9%)、Ruminococcaceae_NK4A214_group(2.8%)和志贺氏埃希菌属(2.0%),虾肠道中的优势菌群为盐单胞菌属(3.1%)、弧菌属(1.1%)和志贺氏埃希菌属(1.1%)。
2.4 微生物的群落相似性
对2种模式的不同样本微生物进行聚类分析,由图4可知,发现同一样本的微生物群落先聚集在一起,不同模式中水样和底泥样本的微生物群落结构具有相似性,而混养的虾肠道微生物群落先和其他样本聚集在一起,說明虾肠道中的微生物群落结构在2个模式中存在一定差异。
2.5 2种养殖模式下的水体微生物标志物
通过LEfSe分析组间菌群差异,可以找出各组间特异的主要菌群,由图5可知,主养池塘的微生物标志物共有8个类群,分别为双歧杆菌目(Bifidobacteriales)、双歧杆菌科(Bifidobacteriaceae)、双歧杆菌属(Bifidobacterium)、Ruminiclostridium_5、Fusicatenibacter、Subdollgranulum、EF100_94H03和Pedobacter。混养池塘的微生物标志物共有18个类群,分别为Bacteroides_plebeius_DSM_17135、蓝色芽殖杆菌属(Gemmobacter)及其中的CC_PW_75种、外硫红螺菌科(Ectothiorhodospiraceae)、外硫红螺菌属(Ectothiorhodospira)及其中的enrichment_culture种、Candidatus_Planktoluna、alphal_cluster、柯克斯体属(Coxiella)、Oligoflexus、真杆菌科(Eubacteriaceae)、博斯氏菌属(Bosea)、甲基孢囊菌属(Methylocystis)、Alpinimonas、拉恩氏菌属(Runella)、Oligoflexaceae、束缚杆菌属(Haliscomenobacter)和12 up。
2.6 不同分类水平上的群落结构
在纲、目、科不同分类水平上,统计丰度>1%的菌群,不同样品种类的优势细菌组成及丰度存在明显不同。由表2可知,在纲分类水平上,放线菌纲(Actinobacteria)是水样中的优势菌群,而且在主养池塘水体(9.3%)中的丰度明显大于其他样品。厌氧绳菌纲是池塘底泥中的优势菌群。梭菌纲是水样和虾肠道的优势菌群,主养池塘的水体(14.1%)和虾肠道(39.6%)丰度明显高于其他样本。拟杆菌纲在水和虾肠道中属于优势菌群,在主养池塘的虾肠道中(40.8%)丰度明显高于其他样本。鞘脂杆菌纲在底泥和水中都是优势菌群,而在虾肠道(<0.1%)中明显低于其他样本。δ-变形菌纲在底泥、水和虾肠道中均属于优势菌群。
在目分类水平上,厌氧绳菌目是池塘底泥中的优势菌群。梭菌目是水样和虾肠道中的优势菌群,主养池塘的水体(14.1%)和虾肠道(39.6%)丰度明显高于其他样本。拟杆菌目在水和虾肠道中属于优势菌群,主养池塘的虾肠道(40.8%)中丰度明显高于其他样本。鞘脂杆菌目在底泥和水中均是优势菌群,而在虾肠道(<0.1%)中明显低于其他样本。气单胞菌目在混养池塘的虾肠道中属于优势菌群。
在科分类水平上,厌氧绳菌科无论是在主养池塘还是套养池塘的底泥中,均是优势菌群。毛螺菌科在水中是优势菌群,其在主养池塘的虾肠道(24.6%)中明显高于混养池塘的虾肠道(<0.1%)。瘤胃球菌科在主养池塘虾肠道中是优势菌群(12.9%)。未分类拟杆菌科Bacteroidales_S24-7_group在主养池塘的虾肠道中(25.5%)丰度明显高于其他样本。气单胞菌科(9.6%)和弧菌科(1.6%)在混养池塘的虾肠道中属于优势菌群,而弧菌科在主养池塘的虾肠道中(1.1%)属于优势菌群。
2.7 主要代谢通路分析
通过KEGG对样品基因序列进行微生物代谢功能分析,由表3可知,在一级水平上,新陈代谢功能模块中相应功能的基因无论是在多样性还是数量上均较多,其次是环境信息加工过程和遗传信息加工过程,细胞过程和未分类模块相应功能的基因数量较少。在二级水平上,基因组中最大的类群是膜运输,其次是复制和修复、翻译、能量代谢、核苷酸代谢和缺乏特征的功能预测,其他如信号传导、转录、氨基酸代谢、酶家族、碳水化合物代谢、细胞运动性也有一定丰度。主养模式在这些代谢通路中基因注释的相对丰度值均高于混养模式。
3 讨论与结论
研究結果显示,在水、底泥和凡纳滨对虾肠道样品中共检测出59 个门、139 个纲、191 个目、348 个科和683 个属。池塘样品的整体多样性水平顺序为底泥>水>虾肠道,与文献中底泥微生物的多样性和丰富度高于水体的结果[6,12]一致。杜世聪等在凡纳滨对虾发病前后的微生物菌群研究中发现,随着疾病的暴发,养殖水体浮游细菌群落α多样性降低[4]。黄雪敏等也发现,养殖水体中更高的α多样性有利于水体环境稳定和虾幼体的健康[13]。本研究中主养塘虾肠道和底泥的多样性水平均高于混养塘,说明主养塘整体的微生态环境要好于混养塘。
王一亭等认为,黄颡鱼、草鱼、鲢、鳙混养模式下的水体微生物群落多样性高于单养模式,鲢鳙的引入还可增加水体微生物群落多样性[10]。田相利等发现,草鱼、鲢、鲤混养使养殖环境中微生物的结构组成与代谢功能更加多样化[14]。而本研究中,主养池塘(单养模式)的整体多样性水平要高于混养池塘(混养模式),说明添加的有益菌对池塘微生物群体的多样性起到了非常良好的作用,有效促进了凡纳滨对虾主养池塘的菌群多样性水平,保持了系统良好的物质和能量流动。
在内陆湖泊、水库等淡水自然生态系统中,底泥细菌群落结构都相对稳定,变形菌门、拟杆菌门和绿弯菌门往往是主要的优势菌群[15-16],凡纳滨对虾主养和混养2种模式中底泥的优势菌群都是变形菌门、拟杆菌门、绿弯菌门和酸杆菌门,其中混养池塘中主养品种为草鱼,这与章海鑫等主养草鱼池塘底泥中的优势菌门结果[8]一致,与文献中凡纳滨对虾、 斑点叉尾和暗纹东方鲀养殖池塘的底泥群落结构[5,7,9]类似。
Zwart等发现,蓝细菌门和拟杆菌门也是不同淡水环境(湖泊和河流)常见的优势菌门[17]。本研究中,水体的优势菌群为变形菌门、拟杆菌门、厚壁菌门和蓝菌门,与文献中的结果[3-4,9]类似,蓝菌门菌群在不同模式的水体中丰度均较高。蓝细菌门是生态系统中碳循环的主要参与者和初级生产力的主要贡献者,参与氮的固定与修复,提升稻田肥力,在氮、磷丰富且氮磷比例失调的水体中生长旺盛,可作为水体富营养化的指示生物,大量繁殖易造成水资源污染[18-20],凡纳滨对虾2种模式水体中蓝细菌门丰度较高,说明水体有一定程度的富营养化。
当对虾养殖到一定程度,虾肠道细菌区系会相对稳定,优势群落相对固定[21],变形菌门在不同模式虾肠道的样品中都是优势菌群,这个结果与克氏原螯虾(Procambarus clarkii)、凡纳滨对虾、罗氏沼虾(Macrobrachium rosenbergii)等甲壳类动物的研究结果[19,22-24]一致。变形菌门是甲壳类水生无脊椎动物肠道微生物的优势种,是肠道菌群失调的微生物标志[25]。本研究中,虾肠道的优势菌群除了变形菌门外,主养池塘中还有拟杆菌门和厚壁菌门,混养池塘中还有蓝菌门和软壁菌门。主养池塘的结果与文献结果[5,26-28]类似,而混养池塘中,由于受到鱼类的影响,混养池塘优势菌群的结构组成和单养凡纳滨对虾的池塘有一定的差别,这也说明了虾肠道的微生物菌群组成受到环境的影响。
拟杆菌门为化能异养细菌,可以有效利用水体中的溶解性有机物[29],与DNA、蛋白质及脂类等有机物的转换有密切联系,没有被虾完全消化的饲料会不断随粪便排入水体,造成水体中拟杆菌门的堆积[30]。王一亭等认为,水体及底泥中拟杆菌门细菌增加,是因为草鱼饵料残渣和粪便沉积在水体和底泥中,导致拟杆菌门细菌大量繁殖[10]。本研究水体和底泥中拟杆菌门含量均较高,与文献中的推测一致,可能是由于在养殖过程中虾粪便的堆积,造成水体和底泥中拟杆菌门成为了优势菌群。
金若晨等认为在发病塘的虾肠道中检测到酸杆菌门和硝化螺旋菌门,暗示了水体和底泥环境的变化影响了凡纳滨对虾的健康[5]。本研究中酸杆菌门和硝化螺旋菌门都是底泥中的优势菌群,而在水中和虾肠道中仅检测到很少的含量(硝化螺旋菌门<0.1,酸杆菌门<0.9),与文献结果[31]一致。酸杆菌门是一种嗜酸菌,是苯酚的主要降解菌[32],有研究认为,酸杆菌门对纤维素有一定的分解能力[33],草鱼粪便中大量的纤维素是造成底泥样本中酸杆菌门占比较高的原因[10]。混养池塘中主养品种为草鱼,而主养池塘上一季养殖的品种也是草鱼,这也许是2个池塘底泥中酸杆菌门含量高的原因。硝化螺旋菌门是一类革兰氏阴性菌,能减少污水中铵离子的含量,维持氨氮、硝酸盐和亚硝酸盐等氮循环体系[34],在亚硝酸盐转化至硝酸盐的过程中发挥着重要作用[35]。
变形菌门包括一些病原菌,如大肠杆菌、假单胞菌和弧菌等,变形菌门在肠道所占丰度的提高可能反映了肠道菌群的失衡[36]。气单胞菌目、气单胞菌科、气单胞菌属、弧菌科、弧菌属的微生物菌群在混养池塘的虾肠道中属于优势菌群,弧菌科、弧菌属在主养池塘的虾肠道中属于优势菌群。弧菌是水产养殖中常见的条件致病菌,发病对虾肠道中弧菌含量显著高于健康对虾[37],是引起对虾白便综合征[38]的主要致病菌,是虾幼苗时期的主要致病菌,能导致虾死亡[39]。杜世聪等在凡纳滨对虾发病前后的微生物菌群研究中发现,随着疾病的暴发,细菌群落结构更加分散,标志着类群种间互作关系变得更加复杂和无规律[4]。本研究中,主养池塘的微生物标志物共有8个类群,而混养池塘的微生物标志物有18个类群,混养池塘微生物菌群较主养池塘更为无序,而池塘中对虾肠道弧菌含量也较多,但虾本身没有发病,应该属于疾病早期。2个池塘均是带病而未发病,主养池塘中可能是由于添加的益生菌改善了整体的生态环境,而混养池塘则是一种混合、立体式的养殖模式,微生物菌群非常多样化。邱楚雯等认为凶猛的肉食性鱼类会捕食凡纳滨对虾,仅有体质健壮的对虾个体才能够生存[9]。本研究中也有可能是发病的虾被鲤鱼吃掉了,因而没有被发现,同时也从侧面印证了混养的虾没有出现大规模的发病现象。
鞘脂杆菌纲(Sphingobacteriia)、鞘脂杆菌目(Sphingobacteriales)在底泥和水中都是优势菌群,而在虾肠道中(<0.1%)的豐度明显低于其他样本,与文献结果[5]一致。鞘脂杆菌纲与水体中的蛋白质、氨基酸和脂类等分解相关,养殖过程中产生残饵、粪便,这些有机质的分解可能使拟杆菌门含量增多[40]。
本研究底泥样本中属水平上的优势菌群为硫杆菌属(3.2%),水样中属水平上的优势菌群为hgcI_clade(4.9%)。王瑞宁等发现,鳗鲡养殖池塘水体中检测到属水平上的主要优势菌群为hgcI_clade[6]。金若晨等发现,hgcI_clade在水中平均相对丰度为 9.13%,显著高于底泥(0.07%)和虾肠道(1.75%)[5],hgcI_clade是一种可有效利用多种碳水化合物的有益菌属[41]。硫杆菌属细菌可用来去除污水污泥及河流沉积物中的重金属[42],底泥中的硫杆菌属丰度高,可有效促进系统硫元素循环,减少重金属的富集。
斑点叉尾夏季养殖池塘底泥优势菌群为厌氧蝇菌属[7],厌氧绳菌纲、厌氧绳菌目和厌氧绳菌科无论是在主养池塘还是套养池塘的底泥中,均是优势菌群。底泥中厌氧菌群丰度较大,说明水体的溶氧较低,因此在养殖过程中需要加强换水,保持水体和底泥中的溶氧量,调整底泥的微生物菌群组成,进一步改善底泥的微生物生态环境。
本研究中,α-变形菌纲(Alphaproteobacteria)、β-变形菌纲(Betaproteobacteria)、γ-变形菌纲(Gammaproteobacteria)和δ-变形菌纲(Deltaproteobacteria)在底泥、水和虾肠道中都属于优势菌群,与文献结果[6,40]类似。研究发现,变形菌门中的微生物对氮的去除起作用,α-变形菌纲具有较强的固氮能力,β-变形菌纲经常发现于废水脱氮处理过程,γ-变形菌纲能吸收同化小分子溶解性有机物,对物质循环有促进作用,δ-变形菌纲对氮、磷和有机质的循环有重要作用[43-46]。
微生态制剂能够调节对虾养殖系统微生态平衡、改善对虾免疫系统、控制病原菌和病毒,在水产养殖中的应用越来越广泛[40],主养池塘的微生物标志物有双歧杆菌目、双歧杆菌科、双歧杆菌属。双歧杆菌属对宿主有益,具有抗炎作用[47]。对虾的肠道微生物是维持肠道内环境稳定的重要因素[48],其组成与养殖水体的微生物成分密切相关[49],因而在水体中补充有益菌群,一方面可调节水体的微生物组成,促进水体环境更加健康和稳定,同时有利于凡纳滨对虾肠道微生物的结构和组成,形成更加良好的肠道内环境,有利于对虾的健康成长。
微生物群落的新陈代谢功能对其环境中的养分循环和水体净化起着十分重要的作用[50],本研究通过功能预测,发现新陈代谢、环境信息加工和遗传信息加工等功能中,主养模式的多样性水平均高于混养模式,其中,能量、核苷酸和氨基酸代谢等多种代谢能力,也是主养模式优于混养模式,说明主养模式中添加的有益菌群加速了系统整体的物质循环和能量流动,从而使得主养模式的系统稳定性和能量利用率高于混养模式。
凡纳滨对虾淡水养殖过程中水质的调控尤为重要,养殖户多通过添加益生菌和藻类来对水体的微生态环境进行调控,这也是淡水养殖凡纳滨对虾成功的关键因素。通过对池塘水质的调控,有效提高水体、虾肠道和底泥的微生物多样性,塑造更健康、多元化和功能更强大的微生物群落,提高养殖系统的物质循环能力和能量利用效率,更有利于虾体的健康成长。
参考文献:
[1]裘琼芬,张德民,叶仙森,等. 象山港网箱养殖对近海沉积物细菌群落的影响[J]. 生态学报,2013,33(2):483-491.
[2]Orcutt B N,Sylvan J B,Knab N J,et al. Microbial ecology of the dark ocean above,at,and below the seafloor[J]. Microbiology and Molecular Biology Reviews,2011,75(2):361-422.
[3]陳玲,万韦韬,刘兵,等. 稻虾共作对稻田水体微生物多样性和群落结构的影响[J]. 华中农业大学学报,2022,41(1):141-151.
[4]杜世聪,黄雷,杨坤杰,等. 凡纳滨对虾健康状态分化前后养殖水体浮游细菌群落的比较[J]. 生态学杂志,2019,38(8):2456-2465.
[5]金若晨,江敏,孙世玉,等. 凡纳滨对虾养殖环境及肠道微生物群落特征分析[J]. 水产学报,2020,44(12):2037-2054.
[6]王瑞宁,王淼,黄秋标,等. 基于高通量测序的晒塘前后鳗鲡养殖池塘微生物群落结构差异分析[J]. 农业生物技术学报,2020,28(7):1250-1259.
[7]钟立强,李冰,王明华,等. 斑点叉尾养殖池塘底泥微生物群落结构特征及其影响因素[J]. 中国水产科学,2020,27(8):893-905.
[8]章海鑫,付辉云,张燕萍,等. 精养草鱼池塘底泥微生物群落结构分析[J]. 水产科学,2020,39(4):553-559.
[9]邱楚雯,施永海,王韩信. 暗纹东方鲀不同养殖模式下的微生物群落结构[J]. 水产学报,2021,45(2):209-220.
[10]王一亭,李波,王厚红,等. 基于Miseq测序技术分析黄颡鱼不同养殖模式下池塘微生物群落结构多样性[J]. 水生生物学报,2020,44(4):781-789.
[11]赵晓伟,丁君,窦妍,等. 基于MiSeq测序技术分析红鳍东方鲀养殖环境菌群多样性[J]. 生态学杂志,2015,34(10):2965-2970.
[12]Zeng Y H,Ma Y,Wei C L,et al. Bacterial diversity in various coastal mariculture ponds in Southeast China and in diseased eels as revealed by culture and culture-independent molecular techniques[J]. Aquaculture Research,2010,41(9):e172-e186.
[13]黄雪敏,温崇庆,梁华芳,等. 健康和发病凡纳滨对虾糠虾期育苗池水体的菌群结构比较[J]. 广东海洋大学学报,2018,38(4):27-34.
[14]田相利,郑瑶瑶,柳炳俊,等. 草鱼混养系统细菌数量变动和群落功能多样性研究[J]. 中国海洋大学学报(自然科学版),2012,42(11):19-27.
[15]Bai Y H,Shi Q,Wen D H,et al. Bacterial communities in the sediments of Dianchi Lake,a partitioned eutrophic waterbody in China[J]. PLoS One,2012,7(5):e37796.
[16]Zhang J X,Yang Y Y,Zhao L,et al. Distribution of sediment bacterial and archaeal communities in plateau freshwater lakes[J]. Applied Microbiology and Biotechnology,2015,99(7):3291-3302.
[17]Zwart G,Crump B C,Kamst-van Agterveld M P,et al. Typical freshwater bacteria:an analysis of available 16S rRNA gene sequences from plankton of lakes and rivers[J]. Aquatic Microbial Ecology,2002,28:141-155.
[18]Bekker A,Holland H D,Wang P L,et al. Dating the rise of atmospheric oxygen[J]. Nature,2004,427(6970):117-120.
[19]贾丽娟,王广军,夏 耘,等. 不同地区稻虾综合种养系统的微生物群落结构分析[J]. 水产学报,2023,47(6):069107.
[20]史丽娜,可小丽,刘志刚,等. 罗非鱼-鱼腥草共生养殖池塘沉积物菌群结构与功能特征[J]. 中国农学通报,2015,31(14):64-73.
[21]罗鹏,胡超群,谢珍玉,等. 凡纳滨对虾咸淡水养殖系统内细菌群落组成的PCR-DGGE分析[J]. 热带海洋学报,2006,25(2):49-53.
[22]Shui Y,Guan Z B,Liu G F,et al. Gut microbiota of red swamp crayfish Procambarus clarkii in integrated crayfish-rice cultivation model[J]. AMB Express,2020,10(1):5.
[23]Fan J Q,Chen L M,Mai G Q,et al. Dynamics of the gut microbiota in developmental stages of Litopenaeus vannamei reveal its association with body weight[J]. Scientific Reports,2019,9(1):734.
[24]Ma R R,Wang Y A,Zhao S,et al. The composition of the microbial community associated with Macrobrachium rosenbergii zoeae varies throughout larval development[J]. Journal of Fish Diseases,2020,43(4):413-421.
[25]Holt C C,Bass D,Stentiford G D,et al. Understanding the role of the shrimp gut microbiome in health and disease[J]. Journal of Invertebrate Pathology,2021,186:107387.
[26]郁维娜,戴文芳,陶震,等. 健康与患病凡纳滨对虾肠道菌群结构及功能差异研究[J]. 水产学报,2018,42(3):399-409.
[27]董学兴,吕林兰,赵卫红,等. 不同养殖模式下罗氏沼虾肠道菌群结构特征及其与环境因子的关系[J]. 上海海洋大学学报,2019,28(4):501-510.
[28]Tzeng T D,Pao Y Y,Chen P C,et al. Effects of host phylogeny and habitats on gut microbiomes of oriental river prawn (Macrobrachium nipponense)[J]. PLoS One,2015,10(7):e0132860.
[29]Cottrell M T,Kirchman D L. Natural assemblages of marine proteobacteria and members of the Cytophaga-Flavobacter cluster consuming low-and high-molecular-weight dissolved organic matter[J]. Applied and Environmental Microbiology,2000,66(4):1692-1697.
[30]Michaud L,Lo Giudice A,Troussellier M,et al. Phylogenetic characterization of the heterotrophic bacterial communities inhabiting a marine recirculating aquaculture system[J]. Journal of Applied Microbiology,2009,107(6):1935-1946.
[31]裴鹏兵,吴洁琼,梁宏豪,等. 生物净水栅对凡纳滨对虾肠道菌群组成的影响[J]. 水产科学,2018,37(3):301-308.
[32]石焱,马徐发,吴志新,等. 水蕹菜浮床养殖模式下黄颡鱼肠道菌群结构分析[J]. 华中农业大学学报,2016,35(2):69-76.
[33]傅丽君,安新丽,郑天凌. 环境中放线菌及其抑藻活性物质研究的若干进展[J]. 地球科学进展,2010,25(9):960-965.
[34]房昀昊,彭剑峰,宋永会,等. 高通量测序法表征潜流人工湿地中不同植物根际细菌群落特征[J]. 环境科学学报,2018,38(3):911-918.
[35]王皓,钱琪卉,丁瑞睿,等. 复合潜流人工湿地对农村生活污水的净化效果及其微生物群落结构特征[J]. 安徽农业大学学报,2020,47(6):962-970.
[36]Shin N R,Whon T W,Bae J W. Proteobacteria:microbial signature of dysbiosis in gut microbiota[J]. Trends in Biotechnology,2015,33(9):496-503.
[37]杨坤杰,王欣,熊金波,等. 健康和患病凡纳滨对虾幼虾消化道菌群结构的比较[J]. 水产学报,2016,40(11):1765-1773.
[38]曹海鹏,温乐夫,周桂娴,等. 南美白对虾白便综合征病原霍乱弧菌的分离与药敏试验[J]. 动物医学进展,2016,37(2):128-132.
[39]Zhou J F,Fang W H,Yang X L,et al. A nonluminescent and highly virulent Vibrio harveyi strain is associated with bacterial white tail disease of Litopenaeus vannamei shrimp[J]. PLoS One,2012,7(2):e29961.
[40]刘洋,宋志文,李凌志,等. 微生态制剂-生物膜对虾养殖系统水质净化效果研究[J]. 水生态学杂志,2020,41(1):92-99.
[41]Ghylin T W,Garcia S L,Moya F,et al. Comparative single-cell genomics reveals potential ecological niches for the freshwater acI Actinobacteria lineage[J]. The ISME Journal,2014,8(12):2503-2516.
[42]高杰. 微生物在污泥無害化处理中的应用[J]. 绿色科技,2018(10):117-118.
[43]Liang Y H,Li D,Zhang X J,et al. Microbial characteristics and nitrogen removal of simultaneous partial nitrification,anammox and denitrification (SNAD) process treating low C/N ratio sewage[J]. Bioresource Technology,2014,169:103-109.
[44]王鹏,陈波,张华.基于高通量测序的鄱阳湖典型湿地土壤细菌群落特征分析[J]. 生态学报,2017,37(5):1650-1658.
[45]Naganuma T,Fukai I,Murakami Y,et al. Effect of ultraviolet radiation on the bioavailability of marine diatom-derived low-molecular-weight dissolved organic matter[J]. Aquatic Ecosystem
Health and Management,2000,3(1):163-166.
[46]Lü X F,Yu J B,Fu Y Q,et al. A meta-analysis of the bacterial and archaeal diversity observed in wetland soils [J]. The Scientific World Journal,2014,2014:437684.
[47]蔣曼,姚萍,杨涛,等. 实时荧光定量PCR法研究溃疡性结肠炎患者肠道双歧杆菌属、柔嫩梭菌属及拟杆菌属量的变化[J]. 中国微生态学杂志,2013,25(11):1245-1249,1254.
[48]樊英,王晓璐,李乐,等. 基于高通量测序的不同养殖系统下凡纳滨对虾肠道和水体中微生物的多样性[J]. 广西科学院学报,2017,33(4):261-267,273.
[49]沈辉,万夕和,何培民,等. 脊尾白虾肠道微生物菌群结构[J]. 微生物学通报,2015,42(10):1922-1928.
[50]Tanentzap A J,Fitch A,Orland C,et al. Chemical and microbial diversity covary in fresh water to influence ecosystem functioning[J]. Proceedings of the National Academy of Sciences of the United States of America,2019,116(49):24689-24695.
猜你喜欢
江苏农业科学(2017年20期)2017-11-30 14:53:29
江苏农业科学(2017年16期)2017-10-27 14:31:12
湖北农业科学(2017年13期)2017-08-08 05:09:44
中国中药杂志(2017年13期)2017-07-31 08:28:44
山东工业技术(2017年12期)2017-07-06 18:29:27
中国中药杂志(2017年7期)2017-05-26 00:05:40
中国医药导报(2017年9期)2017-05-11 18:44:13
中国中药杂志(2016年24期)2017-04-18 17:42:52
中国中药杂志(2017年4期)2017-03-28 17:06:08
中国中药杂志(2017年3期)2017-03-20 21:11:11
