
染色体微阵列分析技术对16p11.2综合征的诊断及遗传学分析
2024-04-29 07:31韩春晓张玉鑫刘颖文李海波
中国妇幼健康研究 2024年4期
韩春晓,张玉鑫,刘颖文,李海波
(宁波市妇女儿童医院出生缺陷综合防治中心,浙江 宁波 315012)
16号染色体的微缺失和微重复与精神发育障碍中的认知、言语延迟和行为异常有关。16p11.2近端区域由断点4和5定义,包含26个已知基因,其中4个是在线人类孟德尔遗传(online mendelian inheritance in man,OMIM)致病基因,大部分基因在大脑的不同区域表达。这个区域内约600kb的周期性间质缺失,是最常见的拷贝数变异区域[1]。16p11.2(TBX6)近端缺失综合征临床特征包括发育迟缓、认知障碍、语言延迟与障碍、自闭症谱系障碍、面部畸形、癫痫或脑电图异常、肥胖、精神疾病、心脏畸形等,并观察到不完全外显率和异质性[2-3]。目前研究证实16p11.2重复携带者的发育与精神运动迟缓、智力障碍、自闭症谱系、强迫与重复行为和精神分裂症的风险增加。本研究通过对16p11.2综合征患者临床症状、妊娠结局和随访信息的收集分析,旨在探讨其表型和遗传学相关性,丰富16p11.2综合征表型谱,为产前诊断和遗传咨询提供有价值的临床依据。
1 对象与方法
1.1 研究对象
于2019年1月至2022年8月宁波市妇女儿童医院就诊并接受染色体微阵列技术(chromosomal microarray analysis,CMA)检测的9 744例患者,受检者样本包括8 999例产前羊水样本,468例外周血样本和277例流产物样本,将检出16p11.2微缺失/微重复综合征22例患者(20例胎儿、2例患儿)纳入描述性分析。所有孕妇及患儿监护人在CMA检测前进行详尽的知情告知并签署知情同意书,本研究已通过医院伦理委员会审查批准[EC(2020-048)]。
1.2 研究方法
1.2.1 临床指征
①产前诊断指征:孕妇外周血胎儿游离DNA产前筛查异常、血清学筛查高风险、胎儿超声结构畸形、超声软指标异常、胎儿生长受限、高龄妊娠、异常妊娠史等;②患儿受检指征:非已知综合征型神经发育障碍、发育迟缓、智力低下、自闭症、面部体征多发畸形、脏器畸形等。
1.2.2 样本采集
①羊水样本采集:在超声引导下对孕19~24+6周孕妇进行羊膜腔穿刺手术,抽取3个15mL羊水置于无菌管中,其中2管用于细胞培养染色体核型分析,1管用于全基因组DNA提取(亲本验证时抽取父母外周血5mL);②外周血采集:采集患儿及父母外周血5mL。
1.2.3 CMA检测
单核苷酸多态性微阵列分析采用全基因组CytoScantm 750k芯片(美国Affymetrix公司),提取的全基因组DNA进行消化、连接、扩增、纯化、片段化、标记、探针杂交、洗涤和扫描。使用配套ChAS4.0(Chromosome Anaylasis Suite 4.0,Affymetrix)软件对扫描图像结果进行计算分析,参考国际公共数据库包括DGV、ClinGen、ClinVar、DECIPHER、ISCA、OMIM、UCSC等,并结合本实验室内部数据及PubMed文献库,依据美国医学遗传学会对CMA拷贝数变异结果解读指南,评判拷贝数变异的临床意义。
1.2.4 资料收集及统计分析
收集孕妇妊娠期情况及患儿生后情况,包括产前超声、血清学检查、遗传学检查、妊娠结局及患儿发育情况等。胎儿出生后进行定期电话召回与健康随访,收集患儿相关资料,包括病史和家族史。对所得结果数据进行描述性统计分析。
2 结果
2.1 产前诊断指征与患儿表型
颈后透明带扫描(nuchal translucency,NT)及其他超声软指标异常5例,血清学筛查高风险和高龄妊娠各4例,无创产前基因检测(non-invasive prenatal testing,NIPT)异常3例,超声结构异常3例(2例脊柱发育异常,1例泌尿系统异常)。另外,2例因癫痫而就诊患儿,其中1例伴有生长发育迟缓,另外1例有惊厥癫痫家族史,其余未见异常,见表1。
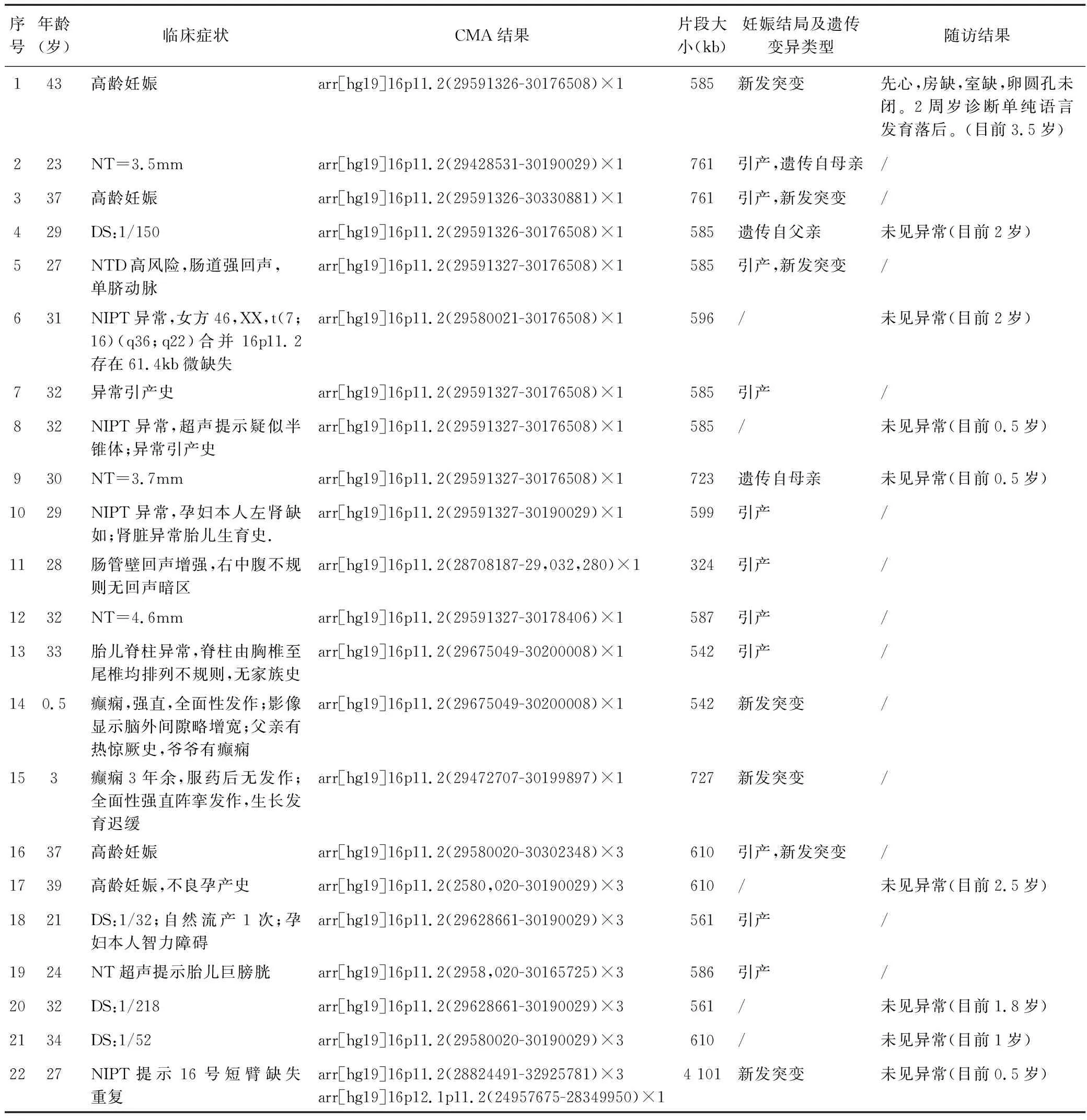
表1 16p11.2综合征患者的特征统计Table 1 Characteristics of patients with 16p11.2 syndrome
2.2 遗传学特征
9 744 例样本中,22例患者(20例产前胎儿、2例患儿)携带16p11.2核心区域拷贝数变异,检出率为0.226%(22/9 744)。在22例拷贝数变异中,21例变异位于近端核心易感区碱基对(base pair,BP)4-BP5,1例变异位于远端核心易感区BP2-BP3。根据变异类型,22例变异中包含15个微缺失变异,占比68.18%(15/22),7个微重复变异,占比31.82%(7/22)。变异片段大小为324~4 101kb。
2.3 变异类型及致病性分级
22例中10例样本进行了亲本验证,7例变异为新发突变,2例变异遗传自母亲,1例变异遗传自父亲。16例胎儿样本进行了染色体核型分析,其中1例样本表现出细胞核型异常,经验证该变异遗传自母亲。根据ACMG致病性评级,22例变异均为致病性变异。
2.4 随访结果
45%(9/20) 孕妇选择继续妊娠。对这9例婴儿进行随访,随访年龄为1.5(0.5~3.5)岁。发现1例缺失携带者婴儿表现异常,出生时诊断为房缺,室缺,卵圆孔未闭,2岁时表现出单纯语言发育落后,经验证发现该变异为新发突变。其余活产婴儿未见明显异常,见表1。
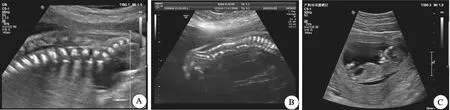
注:A为病例8第一胎,腰骶部椎体排列欠规则,提示半锥体;B为病例13,脊柱排列异常;C为病例19,巨膀胱。图1 胎儿NT超声影像Fig.1 Ultrasonic images of NT of three fetuses
3 讨论
3.1 16p11.2综合征与语言障碍
16p11.2 拷贝数变异最常见的临床症状为语言障碍,占比高达80%~90%。据报道,重复携带者语言延迟与智力障碍相关,而语言障碍在缺失携带者中更普遍[4-5],并未有证据指出语言发育障碍与自闭症谱系有必然联系[6]。文献报道的语言功能障碍多在学龄前(3~7周岁)或学龄期(7~15周岁)确诊[7]。病例1胎儿携带16p11.2近端缺失585kb片段,2岁时被诊断为单纯性语言发育迟缓。其余随访病例尚未发现明显的语言发育落后和自闭症谱系相关症状。这与文献报道的临床占比80%~90%有一定差异,这可能与病例数较少及随访时间较短,患儿表型尚未显现有关,这也是本研究的不足之处。
3.2 16p11.2综合征与心血管系统异常
据统计,0.3%的16p11.2微缺失携带者患有主动脉狭窄、房间隔缺损、卵圆孔未闭等心血管系统异常[8]。Zufferey等在233名16p11.2缺失个体中发现有6%的人患有先天性心脏病[8]。William报道了16p11.2缺失携带者,临床表现为主动脉瓣发育异常、手部畸形、小头畸形等[9]。M Marwan等归纳了16p11.2缺失相关的并发症,包括大头畸形、癫痫、和先天性心脏病[10]。Ghe等报道了携带16p11.2微缺失的同卵双胞胎,分别在11.5岁和13岁时癫痫发病,并出现智力发育迟缓和主动脉瓣发育异常[11]。病例1是本研究中唯一一个心脏畸形的病例,出生时诊断为先天性心脏病,具体表现为房间隔缺损、室间隔缺损、卵圆孔未闭。该缺失携带者片段中包含HIRIP3基因,16p11.2缺失引起HIRIP3基因单倍剂量不足,从而导致主动脉瓣畸形发生,该基因产物与HIRA结合形成的复合物在染色质和蛋白质代谢中发挥着重要作用,表明该基因突变可能为病例1致病因素[11]。
3.3 16p11.2综合征与癫痫
癫痫是16p11.2综合征中高度外显表型[3]。Ben等整理了16p11.2综合征中癫痫发病率,缺失携带者约21.8%~26.8%,重复携带者约19.4%~29%[12]。基因PRRT2相关疾病在16p11.2缺失的个体中已有报道,基因PRRT2与三种常染色体显性疾病相关[12-14]。Natália等认为16p11.2拷贝数变异相关的癫痫复发风险增加,但不具有特异性[1]。本研究报道的病例14,15均因全面性强直性阵挛发作就诊,CMA结果发现两个患者在16p11.2区域分别携带542kb和727kb左右缺失片段,两个片段中均包含致病基因PRRT2,表明该片段缺失可能是癫痫的发病原因。这提示在癫痫患者的遗传学评估时,注意16p11.2微缺失筛查。随访信息中,尚未发现癫痫患者,表明癫痫在该综合征中存在不完全外显率。
3.4 16p11.2综合征与脊柱发育异常
脊柱发育异常是16p11.2复发性综合征产前诊断中最常见的临床表型,占比约21%,具体表现为脊柱侧弯[8]。基因XII和TBX6与常染色体显性和隐性脊髓型肋骨生长障碍5(OMIM 122600)相关,由Chapman等人最早发现TBX6基因在小鼠的前体细胞中参与近轴中胚层的发育[15]。Takemoto等证明了TBX6依赖的SOX2调控轴向干细胞进而影响神经发育[16]。FEI等发现,中国汉族人群中TBX6基因上的2个单核苷酸多态性位点rs2289292和rs3809624在先天性脊柱侧弯的发病中起重要作用[17]。本研究报道了2例胎儿脊柱发育不良,病例8有半锥体发育异常不良孕产史,但未做基因检测,其胎儿携带585kb缺失片段,尚未发现此胎儿脊柱发育异常,未进行亲本来源鉴定,但根据不良孕产史及本次胎儿CMA结果,推测第一胎为16p11.2缺失携带者且变异可能遗传自父(母)亲。病例13超声提示胎儿由胸椎至尾椎排列不规则,CMA提示胎儿携带542kb缺失片段,父母未验证。基于此,并非基因TBX6缺失都出现脊柱发育不良,单纯TBX6基因缺失可能不会发生脊柱发育不良,具体致病机制仍需要进一步的研究探索。
3.5 16p11.2综合征与泌尿系统异常
病例19是16p11.2重复综合征中唯一的结构异常,在颈部透明层(NT)超声检查中提示胎儿为巨膀胱,CMA提示胎儿携带586kb重复片段。在以往报道中16p11.2重复综合征的未发现早期巨膀胱,但J Neurodev等报告了45个16p11.2微缺失和32个微重复,其中3例缺失综合征表现为泌尿系统发育不良,具体表现为肾盂积水、多囊肾病,4例重复综合征表现为肾回流、马蹄型肾[18]。由于病例19终止妊娠,无后续监测,产前标本量不足以进一步检测,未进行单基因变异的遗传学检测,我们推测胎儿在妊娠中后期可能会出现泌尿系统结构或功能异常。泌尿系统异常可能与多种拷贝数变异和单基因突变相关,早期泌尿系统异常可作为微重复综合征的诊断指征仍需更多临床数据支持。另外,病例10孕妇本人左肾缺如,且有肾脏发育不良孕产史,此次胎儿CMA结果提示携带约599kb缺失片段,由于孕妇本人拒绝父母验证,且第一胎未做基因检测,因此无法确定此次胎儿的缺失片段与孕妇肾脏异常及不良孕产史的关系。Li L等报告了一对双胎16p11.2上含有200~240kb缺失区域,双胞胎之一表现为左肾积水[19]。Masuno等报道了一个4月龄女婴,核型为47,XX,+del(16)(p11.2),临床表现为轻微面部异常,肢体异常和泌尿生殖系统异常[20]。Su J等通过CMA分析评估877例肾脏超声异常的胎儿,4例16p11.2缺失携带者中,1例肾脏发育不良,3例单侧肾脏发育不良[21]。
3.6 16p11.2综合征与NT增厚及其他表型
颈部透明层增厚与染色体非整倍体、遗传病和畸形等相关,是行羊水穿刺的产前诊断指征之一。现有研究数据指出NT增厚的异常检出率高达2.5%~10%,单纯与合并其他畸形的NT增厚的致病性拷贝数变异(copy number variants,CNVs)检出率分别为4%和7%[22-23]。本研究中我们共发现3例NT增厚,其中2例(病例2和病例9)遗传自表型正常的母亲,病例9随访时仅为6月龄,尚未见明显异常。因此,NT增厚与16p11.2综合征的相关性还需更多研究数据支持。高龄和血清学筛查高风险并不具有明显的特异性。另外,病例5和病例11超声提示肠道回声增强,相似症状在以往报道文献中已有提及[19,24]。
3.7 小结
本研究不足为样本以羊水居多,超声表型有限,且随访时间较短,病例数较少,未能提供表型外显率以及基因与表型相关性统计学数据。另外,12例变异未明确来源,家系16p11.2复发性拷贝数变异再发风险高于一般人群频率(<1%),再生育前建议对夫妻双方进行CNVs检测及遗传咨询,孕早期需进行有创产前诊断与密切随访。对父母为16p11.2变异携带者家系,应进行遗传咨询告知再生育携带者风险为50%,16p11.2区域缺失和重复外显率分别为46.8%,27.2%[25],建议再次孕育时应行早期的产前诊断或胚胎植入前遗传学诊断以减少出生缺陷患儿发生。
综上所述,16p11.2综合征患者多系统发育表现异常且临床表型多变,异质性强。当临床出现相关表型时,需结合家族史及临床情况并尽早明确遗传学病因。
猜你喜欢
现代临床医学(2022年4期)2022-09-29
河北医学(2021年10期)2021-10-27
中老年保健(2021年12期)2021-08-24
医学与法学(2020年3期)2020-09-18
中国临床医学影像杂志(2019年6期)2019-08-27
现代园艺(2017年21期)2018-01-03
故事作文·高年级(2017年3期)2017-04-12
中国康复理论与实践(2015年10期)2015-12-24
医学研究杂志(2015年5期)2015-06-10
发明与创新(2015年25期)2015-02-27
