
磷化镍修饰磷掺杂氧化镓可见光催化水分解产氢
2024-04-17 00:59:00吕功煊马建泰
无机化学学报 2024年4期
杨 博 吕功煊 马建泰
(1中国科学院兰州化学物理研究所,羰基合成与选择氧化国家重点实验室,兰州 730000)
(2中国科学院大学,北京 100049)
(3兰州大学化学化工学院,兰州 730000)
0 引 言
化石能源的短缺和气候环境的劣化迫使人们寻找新型清洁可再生能源。太阳能具有能量总量大、绿色清洁等优势[1-2]。利用太阳能光催化分解水制氢可以将太阳能转化为清洁的氢能,是一种清洁、绿色、可行的能量转化储存方案[3-4]。含有d10电子结构Ga 元素的氧化镓(Ga2O3)半导体材料的禁带宽度为4.9 eV,具有独特的物理化学特性,被广泛应用于电子[5]、催化[6]、气体传感器[7]和太阳能电池[8]等领域。然而,Ga2O3过宽的禁带宽度导致其只能吸收紫外光,整体太阳能利用效率较低。科学家们提出了多种策略来扩展宽禁带半导体的光吸收范围。构建异质结构是一种提升材料光吸收的重要方法[9-12]。外来元素掺杂也是一种重要的可以有效降低能带宽度的方法[13-15]。例如,N 掺杂TiO2形成TiO2-xNx,半导体的光响应范围从320 nm 扩展至490 nm[16]。Ni 掺杂InTaO4使得NiOy/In0.9Ni0.1TaO4材料的光响应范围扩展至600 nm[17]。在众多不同种类的非金属元素掺杂(如C、N、S、B 和P)中,P 是一种重要的、可用于掺杂各种不同半导体材料的元素。例如P 元素可掺杂TiO2[18-19]、ZnO[20]、g-C3N4[21-22]和CdS[23-24]等半导体,能够扩展半导体的光响应范围。而且P原子还能够调节光生载流子的动力学特性,延长光生电子-空穴对的寿命[25-26]。
半导体表面构筑助催化剂也能够增强光催化剂的光吸收[27-28],而且助催化剂能够接受光激发转移至表面的电子,加快光生电子-空穴对的分离[29-30]。助催化剂还能进一步降低表面反应过电势,进而加快整个光催化反应过程。贵金属如Pt、Ir等是最重要的助催化剂,但贵金属在自然界稀少的含量、昂贵的价格限制了其大范围应用。利用具有类贵金属催化活性的过渡金属磷化物(如CoP、Ni2P、FeP 和Cu3P)与各类半导体材料结合可实现高转化数和效率的光催化制氢[31-36]。通过一步磷化法制备过渡金属磷化物助催化剂修饰P 元素掺杂Ga2O3半导体,有望实现高效的可见光催化水分解,但目前为止这一工作仍未有报道。同时,对过渡金属磷化物作助催化剂的光催化剂在光催化纯水分解中稳定性研究较少[37-39]。
利用含Ni2+溶液浸渍Ga2O3半导体,接着以一水合次亚磷酸钠(NaH2PO2·H2O)为磷源低温磷化法处理得到Ni2P 助催化剂修饰P 元素掺杂Ga2O3的催化剂。通过调节Ni2+负载量和磷源前驱体NaH2PO2·H2O 的加入量对催化剂的活性进行优化,进一步提升了催化剂的活性和稳定性。
1 实验部分
1.1 实验原料
Ga2O3购自天津丰越化学品试剂有限公司;六水合氯化镍(NiCl2·6H2O)、NaH2PO2·H2O 购自天津科密欧化学试剂有限公司;无水硫酸钠(Na2SO4)购自西陇科学;无水甲醇(CH3OH)、无水乙醇(C2H6O)购自成都科隆化学品试剂公司;氯铂酸六水合物(H2PtCl6·6H2O)购自Sigma-Aldrich公司。
1.2 材料制备
1.2.1 Ga2O3-P的制备
将Ga2O3(374.8 mg,2 mmol)和NaH2PO2·H2O(1 271.8 mg,12 mmol)在研钵中混合研磨后置于瓷舟中。瓷舟置于石英管中央,在通Ar 气保护下,150 min 升温至300 ℃,恒温120 min 后,自然降温至室温。所得产物在去离子水、乙醇中洗涤后于50 ℃真空干燥得到橙红色Ga2O3-P产物。
1.2.2x-Ni2P/Ga2O3-P6的制备
分别将不同质量的NiCl2·6H2O(4.8、9.7、14.7、24.2、35.6、52.7 和118.8 mg)溶解于40 mL 无水甲醇中得到含Ni2+离子的浸渍液。Ga2O3(374.8 mg,2 mmol)分散至上述含有Ni2+溶剂中,搅拌30 min,超声30 min 后蒸发除去其中溶剂甲醇。将浸渍不同Ni2+含量的Ga2O3与NaH2PO2·H2O(1 271.8 mg,12 mmol)混合研磨后转移至瓷舟中,在Ar 气保护条件下,150 min 升温至300 ℃,恒温120 min 后自然降温至室温。所得产物经过洗涤、干燥后制得x-Ni2P/Ga2O3-P6产物(其中x代表Ni2+和Ga2O3的物质的量之比,x=1%、2%、3%、5%、7%、10%、20%)。
1.2.3 5%-Ni2P/Ga2O3-Py的制备
XING Peng-fei, LI Zi-fu, LI Qiang, ZHAO Rui, FANG Yi-bin, ZHAO Kai-jun, DAI Dong-wei, YANG Peng-fei, ZHANG Yong-wei,LIU Jian-min
NiCl2·6H2O(24.2 mg,0.10 mmol)溶解于40 mL无水甲醇中。Ga2O3(374.8 mg,2 mmol)分散于上述溶液中,搅拌、超声后加热除去甲醇溶剂。将浸渍Ni2+的Ga2O3粉末与不同质量的NaH2PO2·H2O(0、211.9、635.9、1 271.8 和1 907.8 mg)混合研磨后转移至瓷舟,在Ar 气保护下,150 min 升温至300 ℃,恒温120 min 后自然降温至室温。所得产物经去离子水、乙醇洗涤,干燥后得到5%-Ni2P/Ga2O3-Py(其中y代表NaH2PO2·H2O 与Ga2O3的物质的量之比,y=1、3、6、9)产物。当y=0时,得到5%-NiO/Ga2O3样品。
1.2.4 Pt/Ga2O3-P的制备
采用原位光沉积法在Ga2O3-P 表面负载Pt 纳米颗粒。将Ga2O3-P(50 mg)超声分散于150 mL 去离子水中。滴加265 μL H2PtCl6·6H2O(5 mg·L-1)于上述悬浮液中。采用高纯Ar置换后,在光照搅拌下反应3 h,反应过程中Pt 被沉积于Ga2O3-P 表面。反应后的产物经过离心,50 ℃真空干燥后得到Pt/Ga2O3-P产物。
1.3 材料光催化产氢性能测试
采用一个顶端硅胶垫密封的石英瓶(光照面积9.62 cm2)作反应器测定催化剂的光催化活性。将50 mg 合成的催化剂粉末超声分散于150 mL 纯水中,使用Ar 气置换30 min 以除去反应瓶中所含空气。将反应瓶放置在磁力搅拌器上,确保反应瓶与Xe 灯光源(300 W,BJNBet HSX-F/UV)的距离为15 cm,Xe 灯光源上装配420 nm 截止滤光片。每隔相同的时间间隔从体系中抽取一定量的气相产物,用气相色谱仪检测生成H2的量。
表观量子效率(AQE)的测试:采用带通滤光片获得430、460、520、550、600、650、700 和750 nm 波长的单色光,在各个波长下采用辐射光度计测量照射在光催化反应瓶表面的光量子数(FU 100,Si射线检测器,灵敏度,10~50 μmol·m-2·s-1)。根据反应产生H2的量(nH2)与入射光量子数(Nphoton)的比计算反应过程表观量子效率,如式1所示。
1.4 材料光电催化性能测试
采用上海辰华电化学工作站(CHI660E)测试材料的光电性能。在三电极体系中:将10 mg 催化剂分散于1 mL 溶剂中,超声分散后,移取200 μL 悬浮液滴于1 cm×1 cm的ITO玻璃表面,自然晾干后制得工作电极。饱和甘汞电极(SCE)为参比电极,Pt片为对电极。电解质溶液为0.5 mol·L-1的Na2SO4溶液。瞬态光电流在0 V(vs SCE)的条件下测量,采用特定时间间隔隔断Xe 灯光照。材料的电化学阻抗谱(EIS)在起始电压0 V(vs SCE)下测量,频率范围为0.01~100 000 Hz。阴极线扫伏安曲线(LSV)测定的扫速为10 mV·s-1。
1.5 材料的表征
利用X 射线衍射仪(XRD,Smartlab-SE X-ray diffractometer,Rigaku,Cu 靶,λ=0.154 2 nm,工作电压40 kV,电流40 mA,扫描范围10°~90°)表征合成材料的晶体结构。采用场发射扫描电子显微镜(FESEM,JSM-6701,加速电压5 kV)观察样品的表面形貌。采用透射电子显微镜(TEM,Tecnai-G2-TF20,加速电压200 kV)测试样品的形貌和微观结构。使用X 射线光电子能谱(XPS,ESCALAB 250 Xi,AlKα辐射源,结合能采用C1s284.8 eV 校正)分析催化剂表面的元素组成及价态。利用N2吸附-脱附数据得到材料的比表面积和孔性质(ASAP 2020,Micromeritics,液氮77 K)。使用显微共聚焦拉曼光谱仪(LabRAM HR Evolution,激发波长532 nm)表征催化剂的拉曼(Raman)光谱。使用紫外可见分光光度计(UV-2550,Shimadzu)测试材料的紫外可见吸收光谱(UV-Vis)。采用傅里叶变换红外光谱仪(布鲁克,Vertex 70)测试材料的红外光谱(FTIR)。采用Fluoromax-4荧光光谱仪(Horiba Scientific)测试材料的光致荧光(PL)光谱。采用Agilent 6820 气相色谱检测反应体系中的气体种类及含量。采用电感耦合等离子体发射光谱仪(ICP-OES,Agilent 725-ES)检测溶液中离子浓度。
2 结果与讨论
2.1 催化剂的合成与表征
材料的合成采用两步法:(1)Ni2+溶液浸渍Ga2O3制备前驱体,(2)采用NaH2PO2为磷源磷化处理制备x-Ni2P/Ga2O3-Py催化剂。通过调节浸渍时加入Ni2+和磷化时NaH2PO2的用量制备不同的x-Ni2P/Ga2O3-Py催化剂。采用XRD 对所制备的x-Ni2P/Ga2O3-Py的晶体结构进行表征。图1A 为不同Ni2+加入量制备的x-Ni2P/Ga2O3-P6催化剂的XRD 图。图中出现的衍射峰为单斜β-Ga2O3的XRD 峰(PDF No.43-1012)[40-41]。在x-Ni2P/Ga2O3-P6中并未出现属于Ni2P 的XRD 峰,可能原因为Ga2O3表面的Ni2P 含量较低、分散性较高、或者生成的Ni2P 为非晶态等。相似的现象也发生于Ni2P/Al2O3催化剂[42]。图1B 为不同NaH2PO2加入量合成的5%-Ni2P/Ga2O3-Py的XRD 图。不同5%-Ni2P/Ga2O3-Py与单独Ga2O3相比,XRD 峰并未出现明显差异。这表明,P 掺杂未改变Ga2O3的晶体结构,同时没有出现属于P 的XRD 峰的可能原因为掺杂的P为非晶相物质。

图1 (A)Ga2O3、Ga2O3-P和x-Ni2P/Ga2O3-P6的XRD图;(B)5%-NiO/Ga2O3和5%-Ni2P/Ga2O3-Py的XRD图Fig.1 XRD patterns of(A)Ga2O3,Ga2O3-P,and x-Ni2P/Ga2O3-P6;(B)XRD patterns of 5%-NiO/Ga2O3 and 5%-Ni2P/Ga2O3-Py
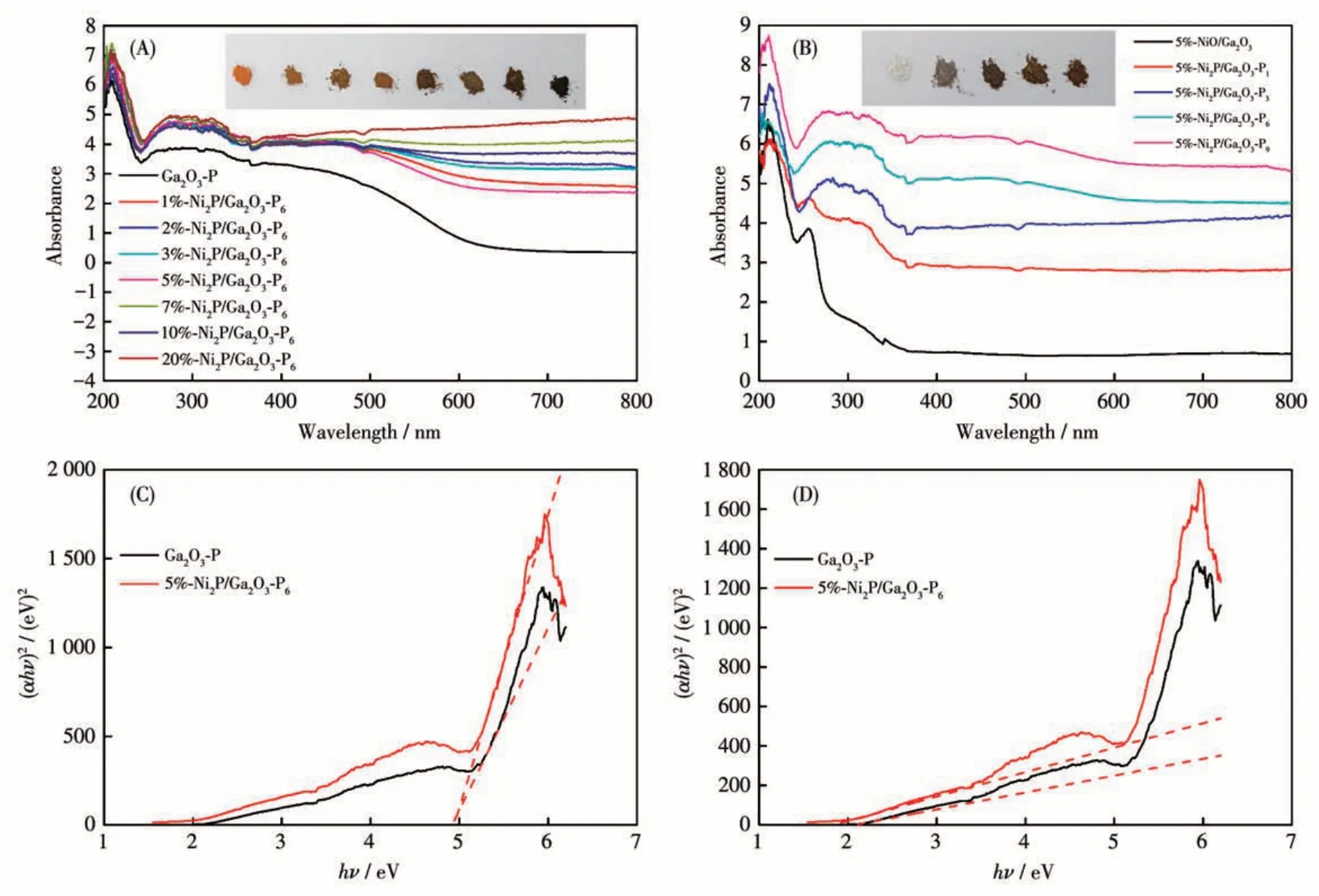
图2 (A)Ga2O3-P和x-Ni2P/Ga2O3-P6、(B)5%-NiO/Ga2O3和5%-Ni2P/Ga2O3-Py的UV-Vis吸收光谱;(C、D)Ga2O3-P和5%-Ni2P/Ga2O3-P6的Tauc曲线Fig.2 UV-Vis absorption spectra of(A)Ga2O3-P and x-Ni2P/Ga2O3-P6,(B)5%-NiO/Ga2O3 and 5%-Ni2P/Ga2O3-Py;(C,D)Tauc plots of Ga2O3-P and 5%-Ni2P/Ga2O3-P6
利用SEM 对合成样品Ga2O3、Ga2O3-P 和5%-Ni2P/Ga2O3-P6的形貌进行表征。Ga2O3的SEM图像显示其为纳米棒状,直径为0.4~1.8 μm,长度为1.33~5.08 μm(图3A)。Ga2O3的更高放大倍数SEM 图中可以观察到其表面存在一定数量的微孔(图3B)[45]。Ga2O3-P 的SEM 图显示磷化后的纳米棒表面变得更粗糙(图3C 和3D)。5%-Ni2P/Ga2O3-P6样品的SEM 图像显示在纳米棒表面出现了明显纳米颗粒状物质,这些纳米颗粒为新形成的Ni2P 助催化剂(图3E 和3F)。采用EDS 能谱对5%-Ni2P/Ga2O3-P6样品中元素含量进行测定(图3G),可以看出样品中含有Ga、O、Ni和P元素,且其中Ga、O、Ni和P元素的原子百分比分别为46.40%、38.59%、0.41%和14.40%。SEM 元素面扫结果证明Ga、O、Ni和P元素在5%-Ni2P/Ga2O3-P6催化剂表面分布均匀(图3H~3L)。

图3 (A、B)Ga2O3、(C、D)Ga2O3-P和(E、F)5%-Ni2P/Ga2O3-P6的SEM图像;5%-Ni2P/Ga2O3-P6的(G)EDS能谱、(H)SEM图和(I~L)元素面扫图Fig.3 SEM images of(A,B)Ga2O3,(C,D)Ga2O3-P,and(E,F)5%-Ni2P/Ga2O3-P6;(G)EDS spectrum,(H)SEM image,and(I-L)element mapping images of 5%-Ni2P/Ga2O3-P6
利用TEM 进一步对Ga2O3、Ga2O3-P 和5%-Ni2P/Ga2O3-P6的形貌和微观结构进行表征。TEM 图像显示Ga2O3为棒状结构(图4A),与SEM 结果一致。在Ga2O3的HRTEM中,出现了0.56和0.36 nm的晶格条纹,分别对应于Ga2O3晶体的(001)和(201)晶面(图4B)[46]。Ga2O3-P的TEM图像显示,Ga2O3-P表面出现了丰富的微孔(图4C),推测其是磷化过程中释放PH3气体所致。在Ga2O3-P 的HRTEM 图中,也观察到了属于Ga2O3(001)晶面的晶格条纹(图4D),表明掺杂并未引起Ga2O3的晶体结构变化。图4E 为5%-Ni2P/Ga2O3-P6的TEM 图像,从图中可以看出,在Ga2O3纳米棒的表面和边缘出现了无规则团聚纳米颗粒,应当为Ni2P助催化剂。在5%-Ni2P/Ga2O3-P6的HRTEM 图中,观察到间距为0.20 和0.36 nm 的晶格条纹,其分别对应于助催化剂Ni2P 的(201)晶面和Ga2O3纳米棒的(201)晶面(图4F),证明Ga2O3-P 表面成功修饰了Ni2P助催化剂。
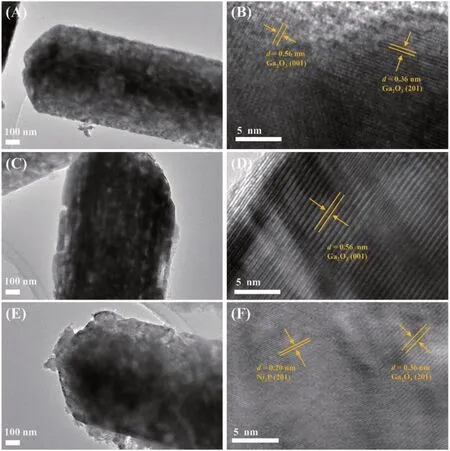
图4 (A)Ga2O3、(C)Ga2O3-P和(E)5%-Ni2P/Ga2O3-P6的TEM图像;(B)Ga2O3、(D)Ga2O3-P和(F)5%-Ni2P/Ga2O3-P6的HRTEM图像Fig.4 TEM images of(A)Ga2O3,(C)Ga2O3-P,and(E)5%-Ni2P/Ga2O3-P6;HRTEM images of(B)Ga2O3,(D)Ga2O3-P,and(F)5%-Ni2P/Ga2O3-P6
利用XPS 表征了催化剂的表面元素组成和化学状态。Ga2O3、Ga2O3-P 和5%-Ni2P/Ga2O3-P6的Ga2p3/2的XPS 峰的结合能分别位于1 117.6、1 118.1和1 118.6 eV(图5A)。Ga2O3、Ga2O3-P 和5%-Ni2P/Ga2O3-P6的Ga3d的XPS 峰的结合能分别位于19.7、20.2 和20.3 eV(图5B)。单独Ga2O3的Ga2p和Ga3d的XPS峰的结合能与文献报道的Ga2O3的值接近[47]。而当Ga2O3被P 掺杂和修饰Ni2P 助催化剂后,其Ga2p3/2和Ga3d的XPS 峰向高结合能方向移动。可能是P 与Ga2O3形成Ga—O—P 键,导致外层的Ga 更缺电子[48-49]。Ga2O3-P 和5%-Ni2P/Ga2O3-P6中的P2p的XPS 谱图如图5C 所示。Ga2O3-P 中的P2p结合能位于129.1、130.0和133.9 eV的峰分别归属于P2p3/2、P2p1/2和表面磷酸盐物种,表明表面存在Pδ-、P0和磷酸盐物种。而在5%-Ni2P/Ga2O3-P6中P2p的结合能为129.5 和130.3 eV 的峰则分别为Ni-P 物种的P2p3/2和P2p1/2[23,50-53]。图5D 为5%-Ni2P/Ga2O3-P6样品的Ni2pXPS 谱图。由于助催化剂Ni2P 的含量较低,Ni2p的XPS 信号较弱。Ni2p中结合能为853.2 和870.9 eV 的峰归属于表面Ni-P 物种,结合能为856.2和873.7 eV 的峰归属于表面部分的NiO 物种,而结合能为861.2和879.3 eV的峰则为卫星峰。
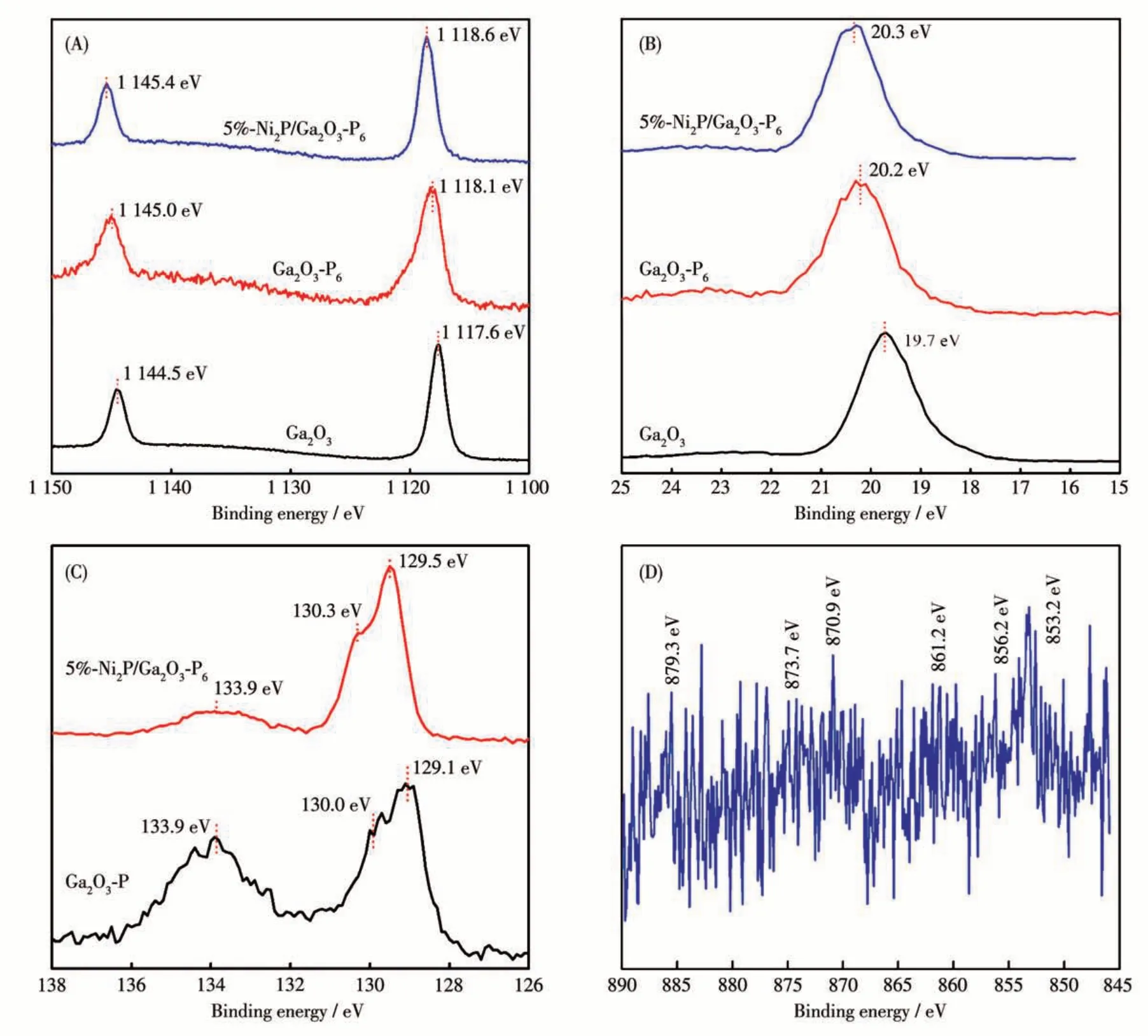
图5 Ga2O3、Ga2O3-P和5%-Ni2P/Ga2O3-P6的(A)Ga2p XPS谱图、(B)Ga3d XPS谱图;(C)Ga2O3-P和5%-Ni2P/Ga2O3-P6的P2p XPS谱图;(D)5%-Ni2P/Ga2O3-P6的Ni2p XPS谱图Fig.5 (A)Ga2p XPS spectra,(B)Ga3d XPS spectra of Ga2O3,Ga2O3-P,and 5%-Ni2P/Ga2O3-P6;(C)P2p XPS spectra of Ga2O3-P,and 5%-Ni2P/Ga2O3-P6;(D)Ni2p XPS spectrum of 5%-Ni2P/Ga2O3-P6
利用Raman 光谱对所制备的催化剂进一步表征,结果如图6A 所示。单斜相Ga2O3的Raman 光谱中出现113.3、146.3、171.9、201.0、350.7、418.4、476.7、654.7 和767.9 cm-1的峰,分别为B1g、B2g、A2g、A3g、A5g、A6g、B4g、B5g和A10g的声子振动频率,与文献报道值比较接近[54]。磷化制备的Ga2O3-P 和5%-Ni2P/Ga2O3-P6中出现了位于356.0、440.0、465.0 cm-1的峰,可以归属为P的A1g、B2g、A2g拉曼活性振动模式[55-57]。
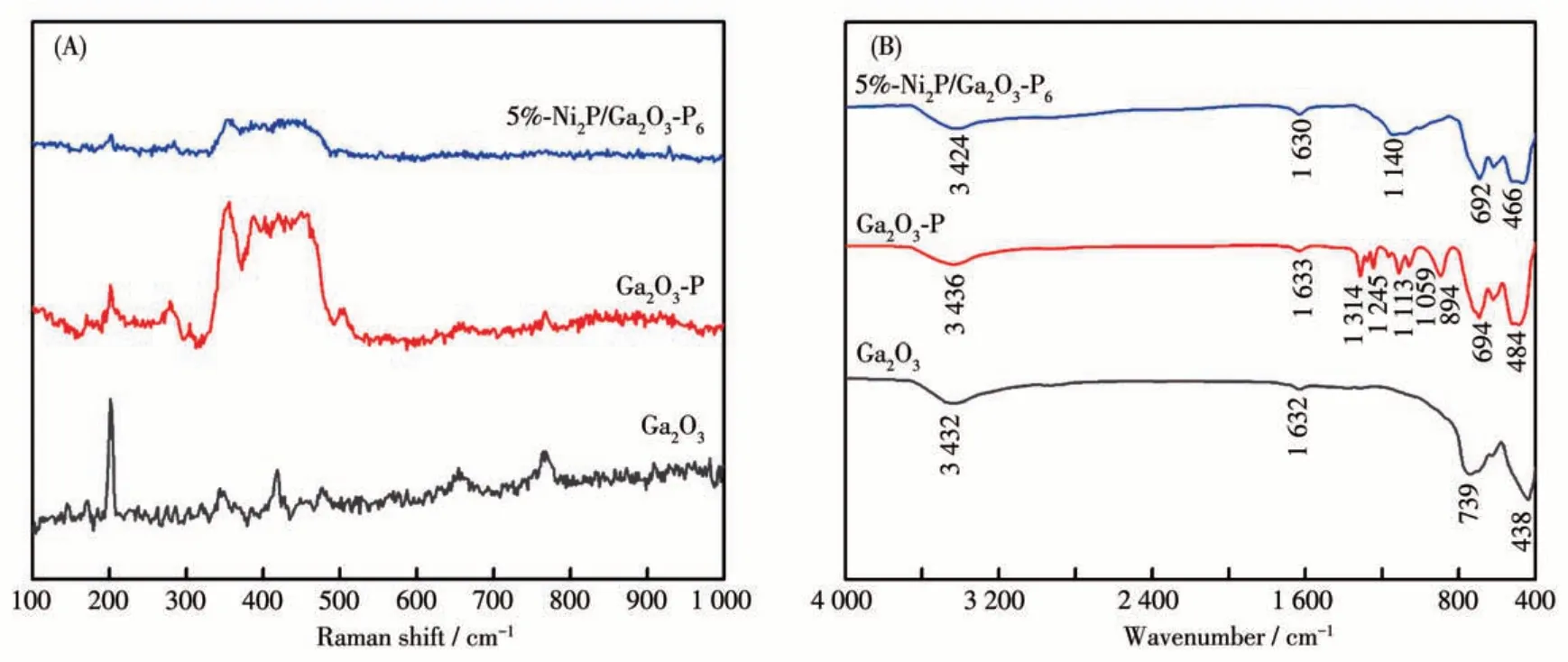
图6 Ga2O3、Ga2O3-P和5%-Ni2P/Ga2O3-P6的(A)Raman谱图和(B)FTIR谱图Fig.6 (A)Raman spectra and(B)FTIR spectra of Ga2O3,Ga2O3-P,and 5%-Ni2P/Ga2O3-P6
图6B 为Ga2O3、Ga2O3-P 和5%-Ni2P/Ga2O3-P6的FTIR 谱图。在Ga2O3的FTIR 谱图中,波数为3 432 cm-1的峰归属为表面羟基的振动形式,而波数为438和739 cm-1处的峰是Ga—O 键的伸缩振动[58-59]。在Ga2O3-P 样品中,由于P 的掺杂使得Ga—O 的伸缩振动峰从739 cm-1位移至694 cm-1,在1 113 和1 245 cm-1波数处出现的峰则分别归属为P—O和P=O的伸缩振动模式[60]。
为了表征材料的比表面积和孔径分布特性,对其进行了N2吸附-脱附等温线的测量。图7A 显示Ga2O3、Ga2O3-P 和5%-Ni2P/Ga2O3-P6的吸附-脱附等温线为存在介孔滞后环的Ⅳ型曲线[61]。表1 显示Ga2O3-P 的比表面积要略大于Ga2O3,这是由于PH3磷化Ga2O3反应过程中引起的扩孔和增大比表面积效应[62]。而当Ga2O3-P 表面修饰Ni2P 助催化剂时,5%-Ni2P/Ga2O3-P6的比表面积略低于Ga2O3-P。图7B显示Ga2O3、Ga2O3-P 和5%-Ni2P/Ga2O3-P6的孔径分布,所有制备样品中孔径分布在介孔范围内,其平均孔径分别为16.18、16.54 和19.68 nm。5%-Ni2P/Ga2O3-P6的平均孔径有所增加是由于引入了Ni2P,形成了接触孔。
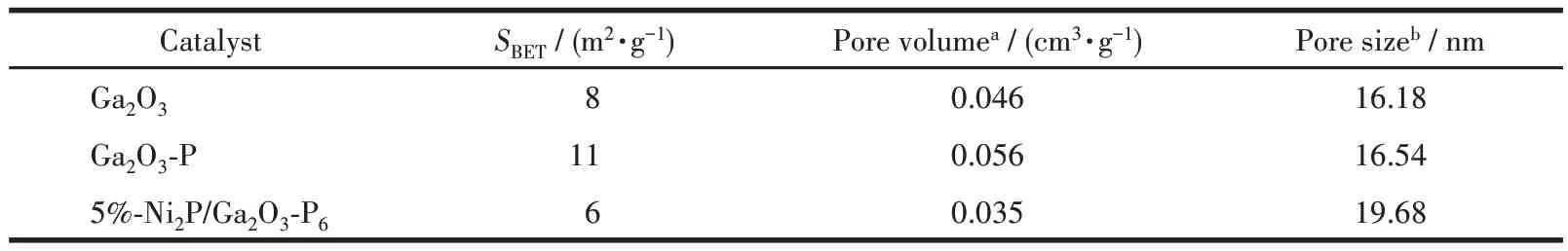
表1 Ga2O3、Ga2O3-P和5%-Ni2P/Ga2O3-P6的比表面积和孔性质Table 1 Surface area and pore properties of Ga2O3,Ga2O3-P and 5%-Ni2P/Ga2O3-P6
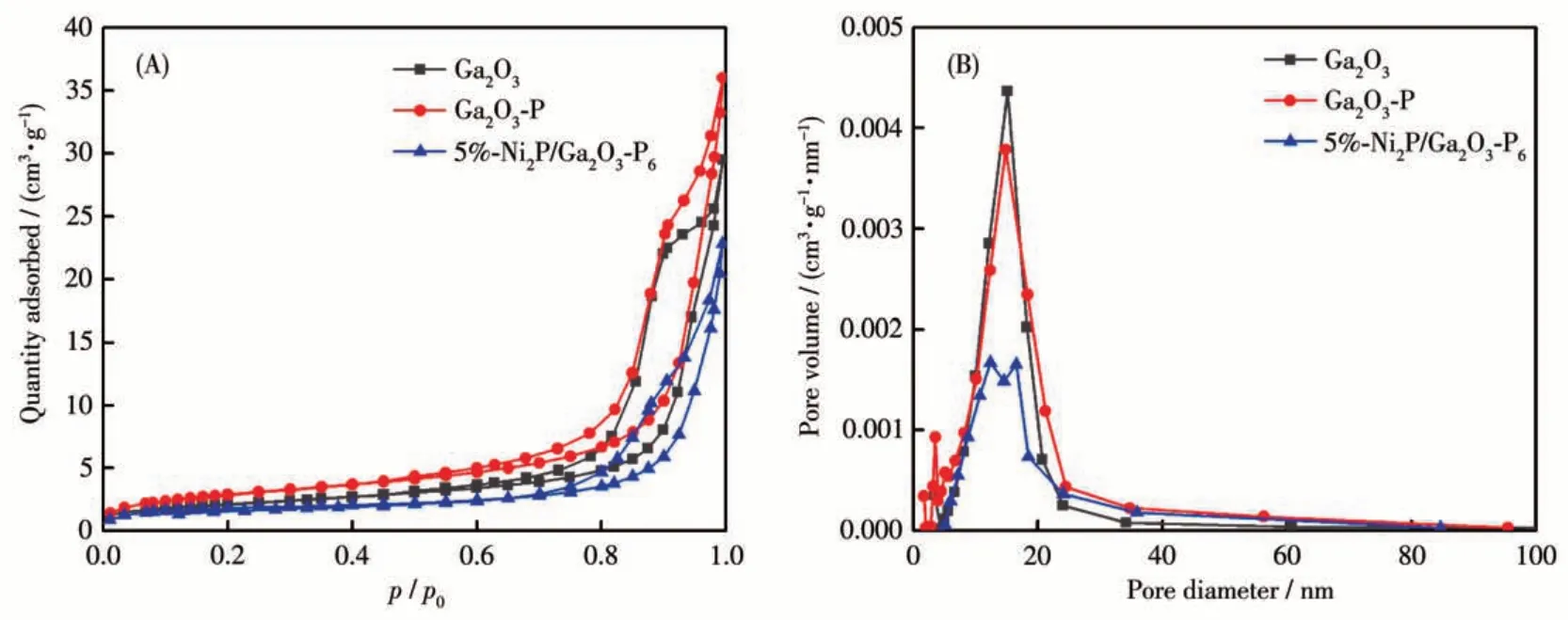
图7 Ga2O3、Ga2O3-P和5%-Ni2P/Ga2O3-P6 的(A)吸附-脱附等温线和(B)Barrett-Joyner-Hallenda(BJH)模型脱附孔径分布曲线Fig.7 (A)Adsorption-desorption isotherms and(B)Barrett-Joyner-Hallenda(BJH)model desorption pore size distribution curves of Ga2O3,Ga2O3-P,and 5%-Ni2P/Ga2O3-P6
2.2 光催化产氢活性
将50 mg 催化剂超声分散于150 mL 去离子水中,Ar 置换后,采用Xe 灯(300 W,截止波长λ>420 nm)为光源照射石英瓶反应器,评价材料的光催化产氢活性。图8A为x-Ni2P/Ga2O3-P6催化剂在光照下的析氢活性对比。Ga2O3-P和x-Ni2P/Ga2O3-P6(x=1%、2%、3%、5%、7%、10%、20%)光催化剂在光照下的平均产氢速率分别为0.13、1.37、1.77、3.66、5.12、3.50、1.90、0.92 μmol·h-1。可以看出,表面负载Ni2P 助催化剂提升了Ga2O3-P的光催化活性。5%-Ni2P/Ga2O3-P6的光催化产氢活性最高,是Ga2O3-P 的39.4 倍,接近于Pt/Ga2O3-P6催化剂的产氢速率(6.20 μmol·h-1)。x-Ni2P/Ga2O3-P6的光催化活性随Ni2P 负载量增长先上升而后下降,在低Ni2P 负载量时,增加Ni2P 的量有助于增加表面反应的活性位点数,促进光催化产氢反应。而当表面Ni2P 量过多时,其对Ga2O3-P 的光吸收有屏蔽作用,不利于光催化反应。图8B 为5%-Ni2P/Ga2O3-Py的光催化产氢活性。NiO/Ga2O3的可见光催化产氢速率为0.026 μmol·h-1,表明NiO/Ga2O3的可见光催化析氢活性较低。随着前驱体磷源加入量的增加,催化剂的光催化活性增加。y为6制备的5%-Ni2P/Ga2O3-P6的光催化产氢速率最高。表明y为6 是较为合适的NaH2PO2·H2O 用量。测量5%-Ni2P/Ga2O3-P6催化剂在不同波长(430~750 nm)单色光照下的量子效率,结果如图8C 所示。可以看出,催化剂在430 nm 波长处的光量子效率最高为0.22%。随着照射光波长的增加,光催化量子效率降低。光量子效率的下降趋势与催化剂的光吸收谱比较好地相匹配,证明材料的光吸收是影响催化剂光量子效率的重要因素。通过光催化循环实验评价Ga2O3-P 和5%-Ni2P/Ga2O3-P6的光催化稳定性,如图8D 所示。可以看出,Ga2O3-P 和5%-Ni2P/Ga2O3-P6的光催化产氢活性随着循环次数的增加而逐渐降低。经过5 次光催化循环反应后,5%-Ni2P/Ga2O3-P6的光催化产氢活性仍为初始值的57.7%。作为对比,我们测试了单独Ga2O3和5%-NiO/Ga2O3催化剂在光催化水分解中的活性和稳定性。纯Ga2O3半导体材料在水溶液中光催化循环中并不能分解水产氢,主要是由于其能带较宽和表面载流子分离效率较低。5%-NiO/Ga2O3光催化剂在全光谱下具有光催化分解水性能,表明NiO 助催化剂也具有捕获光生电子和还原水产氢的能力。但光催化循环实验表明,5%-NiO/Ga2O3材料在光催化反应中也不稳定,其光催化水分解产氢活性随着循环次数的增加而逐渐降低,经过5 次催化循环后的产氢活性是初次的42.8%。采用XRD 对循环反应后的Ga2O3-P 和5%-Ni2P/Ga2O3-P6催化剂的晶体结构进行表征(图9A)。从图中可以看出催化剂在循环反应前后的XRD 衍射峰并未发生明显变化,这是由于样品XRD 图中只出现属于结晶Ga2O3的衍射峰。Ga2O3-P 和5%-Ni2P/Ga2O3-P6循环后的Raman谱图如图9B所示。可以看出,反应后催化剂的Raman 特征峰与反应前相比并无明显不同,表明催化剂的结构在光催化过程中并未发生明显改变。利用HRTEM 对光催化反应后的Ga2O3-P 和5%-Ni2P/Ga2O3-P6催化剂的微观结构进行表征。反应后Ga2O3-P 的HRTEM 中出现的0.36 nm的晶格条纹,归属为单斜Ga2O3的(201)晶面(图9C),与反应前一致。而在结晶Ga2O3外层出现的非晶相层可能为氢氧化镓物种。反应后5%-Ni2P/Ga2O3-P6的HRTEM 中出现晶格间距为0.20 nm 的条纹,为助催化剂Ni2P 的(201)晶面。同时,在结晶Ni2P 的边缘出现了无定型层(图9D)。循环反应后Ga2O3-P 和5%-Ni2P/Ga2O3-P6的XPS 谱图如图10 所示。相比于新制催化剂,循环反应后的Ga2O3-P 和5%-Ni2P/Ga2O3-P6的Ga2p3/2和Ga3d的XPS 峰位移至更高结合能(图10A 和10B),这可能是由于反应后的表面形成了更高价态Ga 的化合物。循环反应后的Ga2O3-P 和5%-Ni2P/Ga2O3-P6的P2p向高结合能方向移动(图10C),这反映催化剂表面的P被部分氧化形成磷酸盐。循环反应后的5%-Ni2P/Ga2O3-P6中的Ni2pXPS 峰也向高结合能方向移动(图10D),这是由于在H2O 环境中,助催化剂Ni2P 表面形成了一层薄的钝化氢氧化镍层,这与反应后催化剂的HRTEM 结果是一致的。活性Ni2P 的表面钝化为氢氧化镍层是光催化剂活性下降的一个重要原因。表2 为Ga2O3-P 和5%-Ni2P/Ga2O3-P6在催化循环反应后溶液中测得的离子浓度。催化剂在光催化过程中发生光氧化,进一步形成离子溶于水介质中是催化剂活性下降的另一个重要原因。

表2 ICP-OES方法测试Ga2O3-P和5%-Ni2P/Ga2O3-P6在光催化循环反应后溶液中Ga3+、PO43-和Ni2+浓度Table 2 Concentration of Ga3+,PO43-,and Ni2+ions in the solution after cycling reaction of Ga2O3-P and 5%-Ni2P/Ga2O3-P6 measured by ICP-OES method
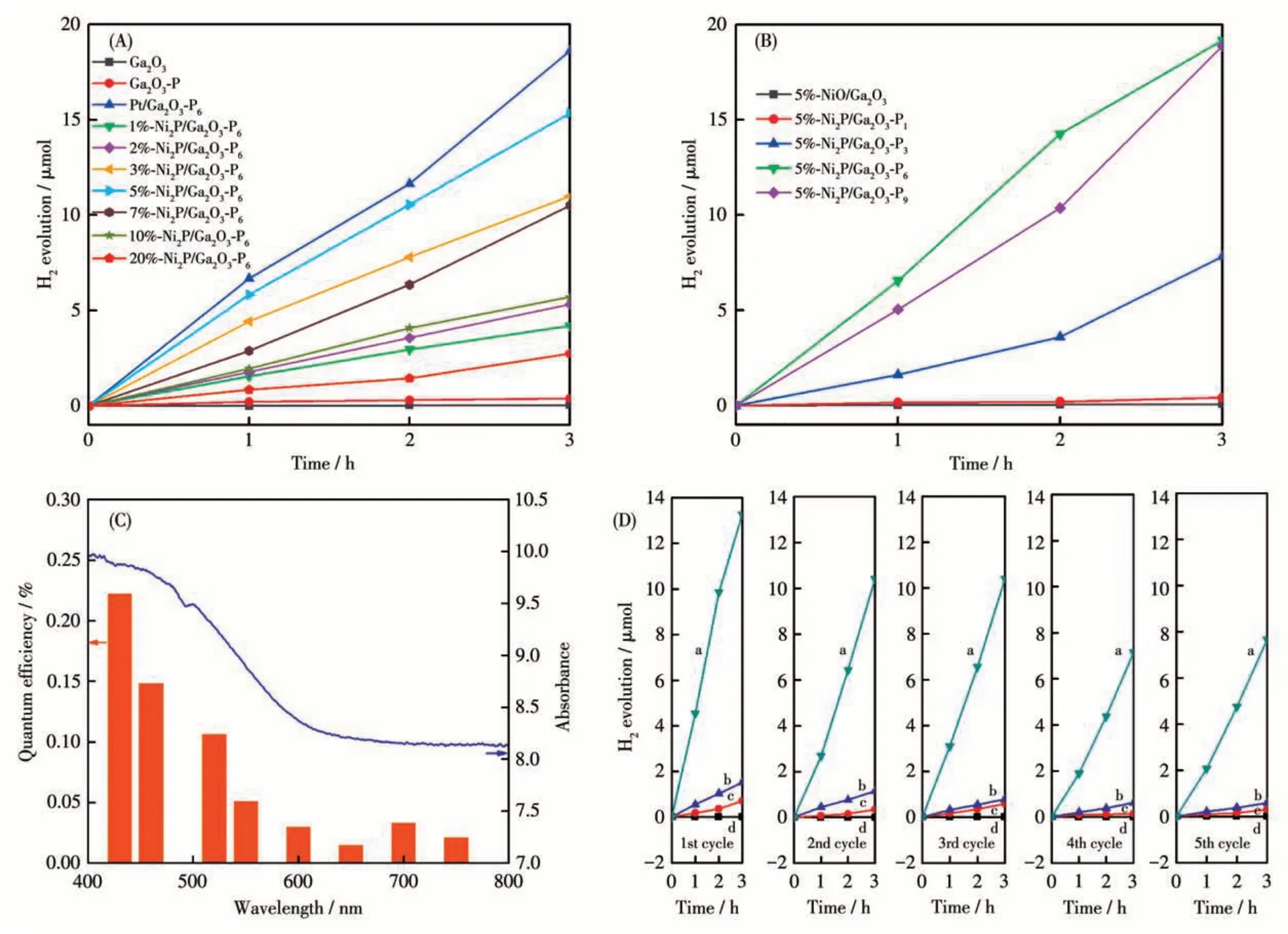
图8 (A)Ga2O3、Ga2O3-P、Pt/Ga2O3-P6和x-Ni2P/Ga2O3-P6的光催化产氢活性;(B)5%-Ni2O/Ga2O3和5%-Ni2P/Ga2O3-Py的光催化产氢活性;(C)5%-Ni2P/Ga2O3-P6的光催化产氢AQE;(D)Ga2O3、5%-NiO/Ga2O3、Ga2O3-P和5%-Ni2P/Ga2O3-P6的长周期循环光催化产氢活性Fig.8 (A)Photocatalytic hydrogen evolution activity of Ga2O3,Ga2O3-P,Pt/Ga2O3-P6,and x-Ni2P/Ga2O3-P6;(B)Photocatalytic hydrogen evolution activity of 5%-Ni2O/Ga2O3 and 5%-Ni2P/Ga2O3-Py;(C)AQE of photocatalytic hydrogen evolution of 5%-Ni2P/Ga2O3-P6;(D)Long term photocatalytic cycling hydrogen evolution activity of Ga2O3,5%-NiO/Ga2O3,Ga2O3-P,and 5%-Ni2P/Ga2O3-P6
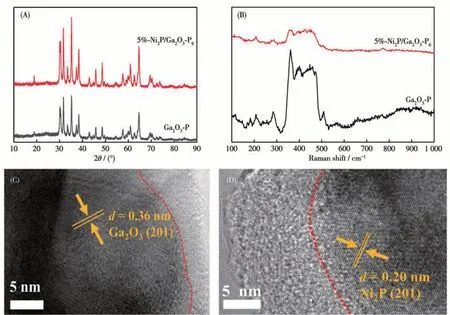
图9 Ga2O3-P和5%-Ni2P/Ga2O3-P6循环反应后的(A)XRD图和(B)Raman谱图;(C)Ga2O3-P和(D)5%-Ni2P/Ga2O3-P6循环反应后的HRTEM图像Fig.9 (A)XRD patterns and(B)Raman spectra of Ga2O3-P and 5%-Ni2P/Ga2O3-P6 after cycling reaction;HRTEM images of(C)Ga2O3-P and(D)5%-Ni2P/Ga2O3-P6 after cycling reaction
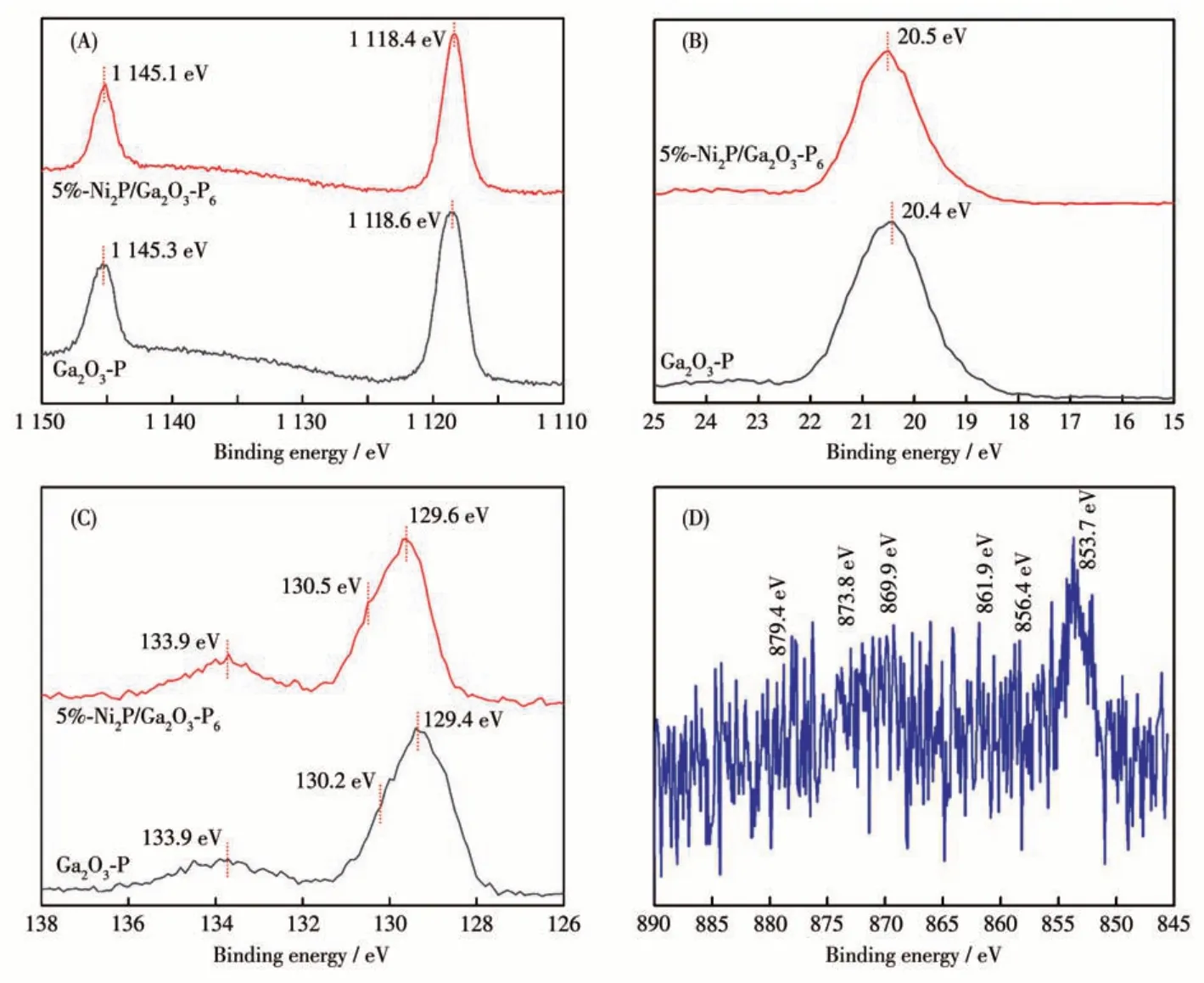
图10 Ga2O3-P和5%-Ni2P/Ga2O3-P6循环反应后的(A)Ga2p、(B)Ga3d和(C)P2p XPS谱图;(D)5%-Ni2P/Ga2O3-P6循环反应后的Ni2p XPS谱图Fig.10 (A)Ga2p,(B)Ga3d and(C)P2p XPS spectra of Ga2O3-P and 5%-Ni2P/Ga2O3-P6 after cycling reaction;(D)Ni2p XPS spectrum of 5%-Ni2P/Ga2O3-P6 after cycling reaction
2.3 催化剂的光电催化性能
为了解释催化剂优良活性的原因,对合成的不同样品进行了光电化学性质研究。Ga2O3、Ga2O3-P和5%-Ni2P/Ga2O3-P6催化剂的光电化学特性曲线如图11 所示。图11A 为Ga2O3、Ga2O3-P 和5%-Ni2P/Ga2O3-P6的瞬态光电流响应(I-t)曲线。相比于纯Ga2O3,Ga2O3-P 的光电流响应值有较大提高,可能原因为掺杂P引起的光吸收增强。助催化剂Ni2P修饰的5%-Ni2P/Ga2O3-P6催化剂的光催化响应电流密度进一步增加,表明Ni2P 能进一步加快半导体表面的电子-空穴分离[21-22]。Ga2O3、Ga2O3-P 和5%-Ni2P/Ga2O3-P6的EIS 如图11B 所示。Ga2O3-P 的奈奎斯特曲线圆弧半径小于Ga2O3,表明电荷在Ga2O3-P 表面传递的阻力小于Ga2O3。P 掺杂增加了Ga2O3的导电能力[63]。当表面进一步负载Ni2P 助催化剂后,5%-Ni2P/Ga2O3-P6的奈奎斯特曲线曲率半径进一步降低,表明电子传递性能进一步增加,这可能是由于Ni2P具有良好的导电性。Ga2O3、Ga2O3-P和5%-Ni2P/Ga2O3-P6的LSV 曲线如图11C 所示。在相同的电势下,Ga2O3-P 具有比Ga2O3更高的阴极极化析氢反应(HER)电流,证明Ga2O3-P 的HER 活性高于纯Ga2O3。表面修饰Ni2P 制备5%-Ni2P/Ga2O3-P6催化剂的HER极化电流密度有更加明显的增加,表明Ni2P 是一个良好的HER 催化剂,Ni2P 助催化剂能够有效降低表面反应过电势[64]。对材料进行PL 表征,进一步反映其表面的光生电子-空穴对的分离迁移效率。图11D 是Ga2O3、Ga2O3-P 和5%-Ni2P/Ga2O3-P6的室温PL光谱。纯Ga2O3的PL 的激发波长为250 nm,可以观察到位于374 nm 的PL 发射光谱,表明Ga2O3具有良好的光致荧光效应[54]。当采用243 nm 的光激发Ga2O3-P6样品时,其PL 谱的峰位于590 nm 处[40]。当Ga2O3-P6表面负载Ni2P 助催化剂时,5%-Ni2P/Ga2O3-P6的荧光峰的强度出现了明显减弱,这是由于Ga2O3-P6表面的光生电子快速向Ni2P 助催化剂转移,表面的载流子复合发光过程受到了抑制[52]。
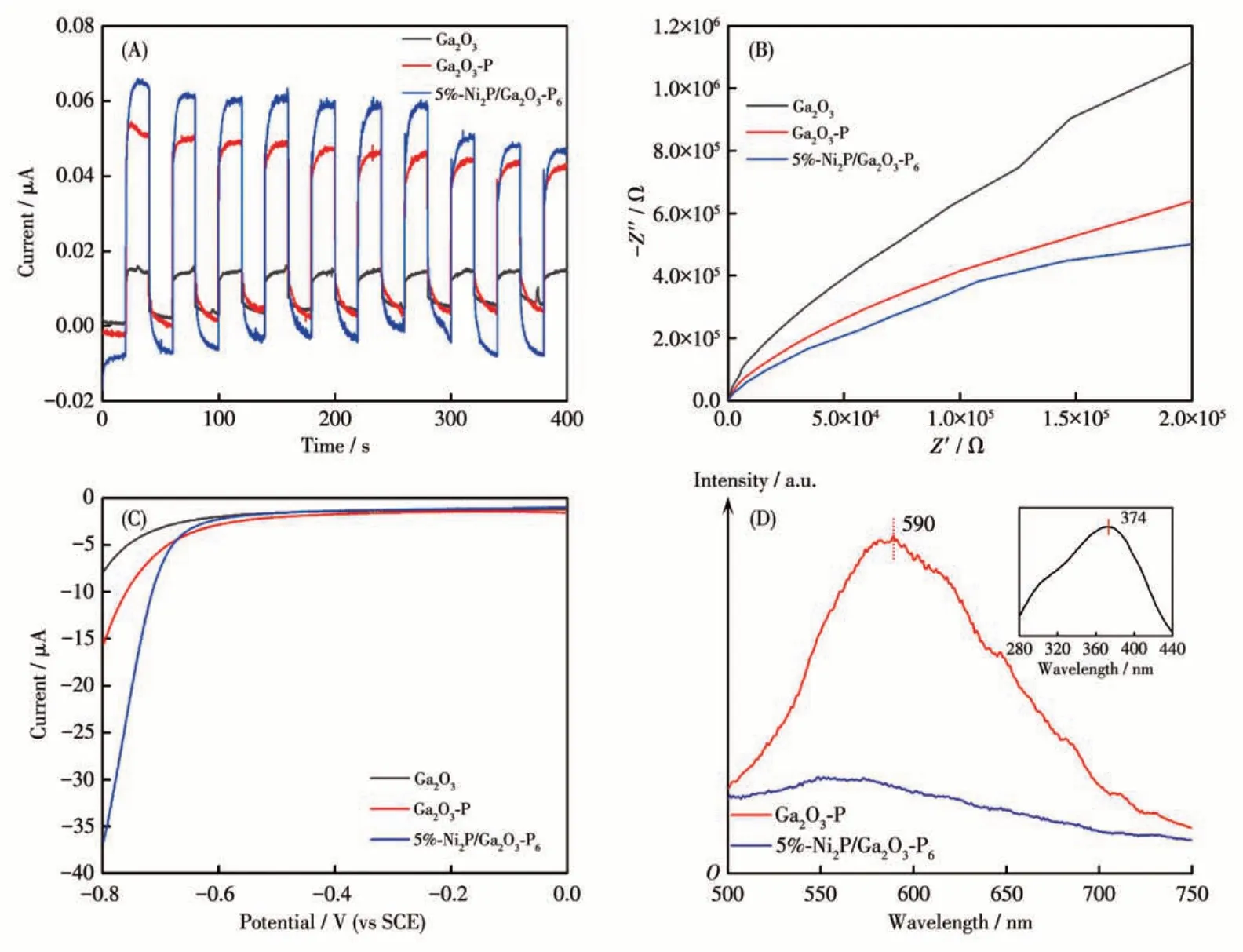
图11 Ga2O3、Ga2O3-P和5%-Ni2P/Ga2O3-P6的(A)I-t曲线、(B)奈奎斯特曲线、(C)LSV曲线和(D)PL谱图Fig.11 (A)I-t curves,(B)Nyquist curves,(C)LSV curves,and(D)PL spectra of Ga2O3,Ga2O3-P,and 5%-Ni2P/Ga2O3-P6
基于前面的表征、光催化产氢活性和光电化学结果,提出了一种可能的光催化水分解产氢的机理(图12)。可见光激发Ga2O3-P6产生光生电子-空穴对。由于合适的能带结构,Ga2O3-P6的光生电子转移至Ni2P 助催化剂,H2O 在Ni2P 表面得到电子被还原为H2。P掺杂Ga2O3起到了两个重要作用:(1)P掺杂扩展了材料的光响应范围,使材料的光响应从紫外光区移至可见区;(2) P 掺杂加速了光生电子-空穴对的分离和转移。Ni2P助催化剂在光催化过程中同样发挥着双重作用:(1)Ni2P 助催化剂能捕获光生电子,加快半导体表面的光生载流子的分离和迁移;(2) Ni2P 助催化剂能降低了产氢反应过电势,加快表面水还原反应动力学速率。
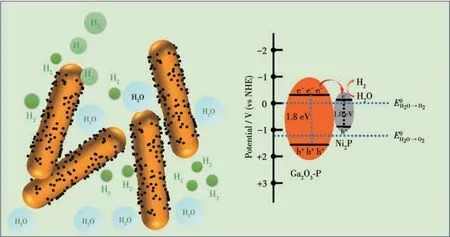
图12 5%-Ni2P/Ga2O3-P6光催化产氢机理Fig.12 Photocatalytic H2 evolution mechanism over 5%-Ni2P/Ga2O3-P6
3 结 论
通过一步低温磷化法制备了Ni2P 修饰的P 掺杂Ga2O3催化剂x-Ni2P/Ga2O3-Py,该催化剂表现出良好的全分解水产氢性能。表面负载助催化剂Ni2P和掺杂P 的量对材料的光催化活性有重要影响。在最优化条件下,5%-Ni2P/Ga2O3-P6的光催化产氢速率达到5.12 μmol·h-1,在430 nm 单色光照射下的量子效率达到0.22%。UV-Vis 吸收光谱、材料光电化学性能测试和PL 光谱证明,在Ga2O3纳米棒中掺杂P 和负载Ni2P 助催化剂扩展了材料的光吸收范围,加速了光生电子-空穴对的分离。长周期光催化性能测试表明,光催化剂表现出良好的稳定性。
猜你喜欢
大学物理(2022年9期)2022-09-28 01:10:52
——潘桂棠光生的地质情怀
沉积与特提斯地质(2021年2期)2021-07-20 06:33:26
物理通报(2020年7期)2020-07-01 09:28:02
无机盐工业(2017年5期)2017-05-25 00:37:34
郑州大学学报(理学版)(2017年1期)2017-04-07 01:09:39
物理化学学报(2017年3期)2017-03-11 00:25:30
化工管理(2017年25期)2017-03-05 23:32:36
昭通学院学报(2016年5期)2016-02-24 10:51:12
原子与分子物理学报(2015年3期)2015-11-24 12:49:34
文理导航(2015年26期)2015-09-29 14:12:24
