
基于UPLC-Q TOF-MSE技术的藤黄健骨丸指纹图谱建立及共有峰鉴定
2024-04-17 07:37:58胡健楠皮子凤戴雨霖
应用化学 2024年3期
谢 东 胡健楠 李 念 杨 桔 黄 青 皮子凤*郑 飞 戴雨霖 越 皓*
(1长春中医药大学吉林省人参科学研究院, 长春 130117)
(2长春人民药业集团有限公司, 长春 130033)
藤黄健骨丸是著名中医学家刘柏龄教授凭借大量的临床实践归纳总结出的中药方剂,该方是由熟地黄、鹿衔草、肉苁蓉、鸡血藤、骨碎补(烫)、淫羊藿和炒莱菔子7 味中药为原料,经粉碎、提取后以炼蜜为粘合剂制成的纯中药浓缩蜜丸制剂[1]。方中7味药,6味入肾经,以补肾治本,治骨治标,标本兼治,现已广泛应用于临床[2-3]。研究表明,该方对骨关节炎、骨质疏松等骨病具有良好的治疗效果[4-5]。然而,目前对藤黄健骨丸的质量标准研究报道较少,赵勇等[6]采用薄层色谱法对方中熟地黄和淫羊藿药材进行了鉴别,利用高效液相色谱(HPLC)技术检测了骨碎补中柚皮苷以及淫羊藿中淫羊藿苷的含量,以提高其质量标准。此外多数相关研究也仅是对方中单个或几个化学成分建立含量测定方法,但检测指标偏少,均难以反映药品的整体质量[7-10]。基于此,本研究采用HPLC 法建立藤黄健骨丸的指纹图谱,结合液质联用技术对其共有峰的结构进行鉴定,并对其相似度进行评价,考察中间体和成品间的相关性,以期为藤黄健骨丸的质量控制提供理论研究基础。
1 实验部分
1.1 仪器和试剂
U3000 型高效液相色谱仪(HPLC,美国Thermo 公司);Acquity 型超高效液相色谱(UPLC,美国Waters 公司);Q-TOF SYNAPT 型高分辨质谱仪(HRMS,美国Waters 公司);ME204/02 型电子天平(上海METTLER TOLED 公司);KS-2200DE型超声波清洗器(昆山洁力美超声仪器有限公司)。
藤黄健骨丸10批中间体(组方中药按制剂工艺制成的混合粉末)批号分别为:22039、22040、22041、22042、23003、23004、23005、23006、23007 和23008,及其对应的10 批成品(组方中药按制剂工艺制成的浓缩蜜丸)批号分别为:20221005、20221101、20221102、20221103、20230301、20230302、20230303、20230401、20230402和20230403,均来源于长春人民药业集团有限公司,药材产地来源分别为: 地黄(河南家种);鹿衔草(内蒙野生);骨碎补(湖北、重庆、贵州野生);肉苁蓉(新疆家种);淫羊藿(甘肃野生);鸡血藤(云南野生);莱菔子(甘肃野生)。对照品松果菊苷(批号:N25GB168967)、淫羊藿苷(批号:T11A11B111118)、柚皮苷(批号:H24N9Z75896)、毛蕊花糖苷(批号:N19GB168679)、淫羊藿次苷Ⅱ(批号:A20GB158231)均购自于上海源叶生物科技有限公司,纯度均大于等于98%。甲醇(色谱纯,美国Tedia公司);乙腈(色谱纯,美国Sigma公司);甲酸(色谱纯,上海Aladdin公司);Milli-Q超纯水系统(美国Millipore公司)。
1.2 对照品溶液的制备
精密称定淫羊藿苷、松果菊苷、淫羊藿次苷Ⅱ、柚皮苷和毛蕊花糖苷对照品各1.0 mg,加甲醇分别配置成浓度为0.1 mg/mL的对照品溶液。
1.3 供试品溶液的制备
精密称定藤黄健骨丸成品5 g 和中间体2.5 g,分别置于50 mL 锥形瓶中,再精密量取甲醇25 mL 加入使溶解,称定质量1,在功率为100 W,频率为40 kHz的条件下超声处理1 h,放冷,称定质量2,用甲醇补足2次称定的质量损失,摇匀,滤过,即得供试样品[11]。
1.4 阴性样品溶液的制备
按照处方工艺制备藤黄健骨丸中7味药材相应的阴性样品,再分别按1.3节方法制备,即得。
1.5 色谱条件
采用Waters Symmetry C18(150 mm×2.1 mm,3.5 µm)色谱柱; 在柱温30 ℃下以0.1%甲酸水(A)和0.1%甲酸乙腈(B)为流动相进行梯度洗脱(0~10 min,5%~10% B; 10~15 min,10%~15% B; 15~30 min,15%~25% B;30~42 min,25%~55% B; 42~52 min,55%~80% B; 52~57 min,80%~5% B; 57~62 min,5% B);流速为0.4 mL/min; 进样量为2 µL; 紫外检测波长为260 nm。
1.6 质谱条件
以ESI 为离子源,在MSE全扫描模式下对正/负离子进行分析; 扫描时间为1.5 s; 扫描范围m/z50~2000; 离子源温度正离子模式下为120 ℃,负离子模式下为110 ℃; 脱溶剂气温度400 ℃; 锥孔气和脱溶剂气的流速为50和800 L/h; 锥孔电压为40 V; 毛细管电压正离子模式设置为3.0 kV,负离子模式设置为2.5 kV; 碰撞能量为25~45 V; 采用甲酸钠对质谱仪进行校正,使用亮氨酸脑啡肽(正离子模式下m/z556.2771,负离子模式下m/z554.2615)对质量数进行实时校正[12]。
2 结果与讨论
2.1 检测波长的选择
通过查阅文献[13-17]确定各对照品常见紫外检测波长,松果菊苷、毛蕊花糖苷的检测波长分别为330 和334 nm,淫羊藿苷的检测波长为270 nm,柚皮苷的检测波长为283 nm,淫羊藿次苷Ⅱ的检测波长为265 nm。结合文献[13-17]并考察藤黄健骨丸紫外检测波长,结果在260 nm 波长下指纹图谱中各共有峰吸收较好,信号较强,故选择其为最适检测波长。
2.2 藤黄健骨丸指纹图谱的建立
2.2.1 仪器精密度试验
取藤黄健骨丸(成品)样品连续进样测定6次,以5号色谱峰(松果菊苷)作为参照峰,计算各共有峰的相对峰面积和相对保留时间[18]。结果见辅助材料表S1和S2,各共有峰的相对峰面积的RSD值均小于1.86%,相对保留时间的RSD值均小于1.52%,表明该仪器精密度良好,满足分析质量要求。
2.2.2 重复性试验
取6份藤黄健骨丸(成品)样品依次进样测定,以5号色谱峰(松果菊苷)为参照峰,计算各共有峰的相对峰面积和相对保留时间[18]。结果见辅助材料表S3 和S4,各共有峰的相对峰面积的RSD 值均小于1.32%,相对保留时间的RSD值均小于1.05%,表明该方法重复性良好。
2.2.3 稳定性实验
取藤黄健骨丸(成品)样品在0、2、4、8、12、16、20和24 h 分别进样测定,以5 号色谱峰(松果菊苷)为参照峰,计算各共有峰的相对峰面积和相对保留时间[18]。结果见辅助材料表S5 和S6,各共有峰的相对峰面积的RSD值均小于1.41%,相对保留时间的RSD值均小于1.22%,说明样品在24 h内稳定性良好。
2.2.4 指纹图谱的建立及共有峰指认
取10批藤黄健骨丸(成品)样品,依次进样测定并记录色谱图,利用“中药色谱指纹图谱相似度评价系统(2012版本)”对供试品溶液的色谱图(X1-X10)进行分析,选择X1作为参照图谱,采用中位数法,时间窗宽度设置为0.1,再进行多点校正以及Mark 峰的匹配,最终获得10 批成品的指纹图谱(图1),并自动生成对照指纹图谱R(图2A谱线a)[19]。因5号峰峰面积大,分离度好,保留时间及峰面积稳定,因此以5号峰为参照峰,共标定出20个共有峰,共有峰面积占总峰面积90%以上。
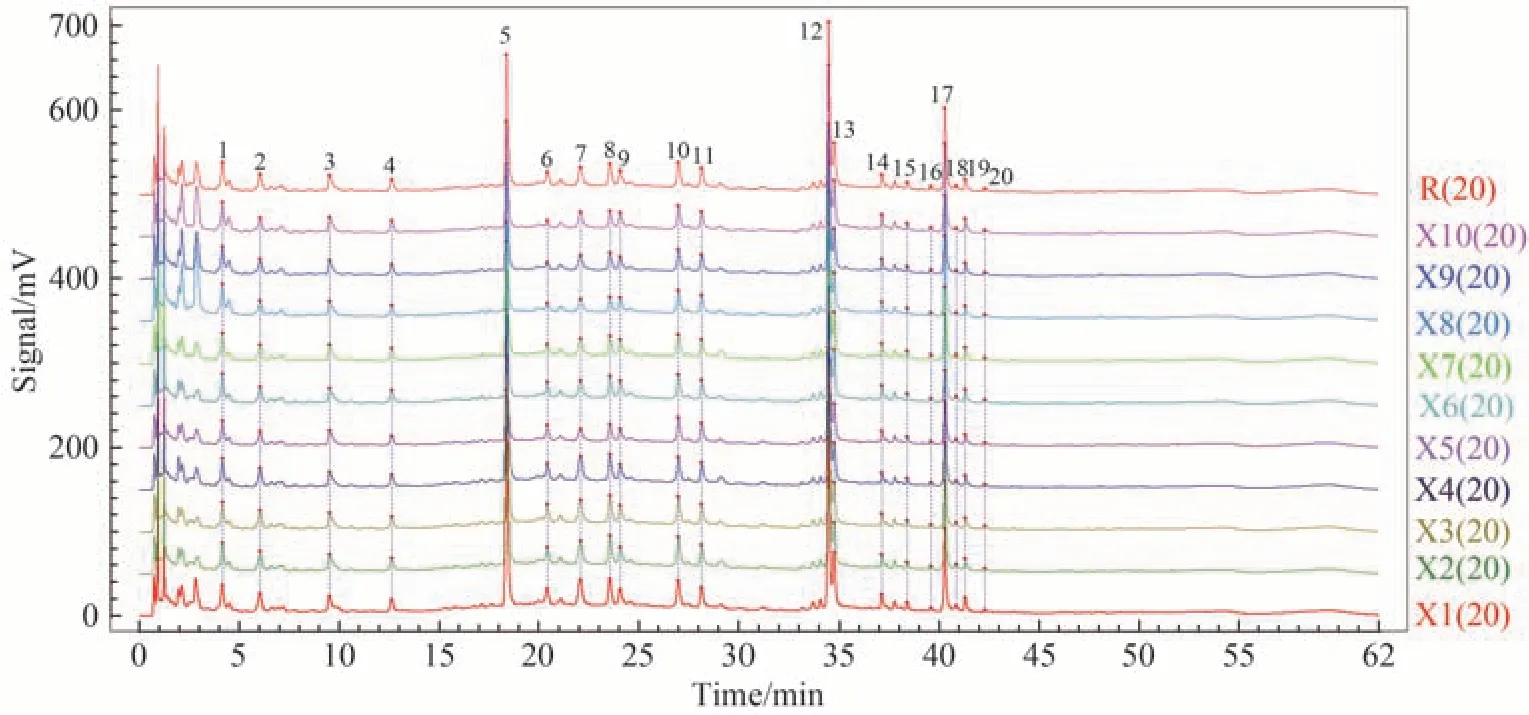
图1 10批藤黄健骨丸指纹图谱Fig.1 Fingerprints of 10 batches of Tenghuang Jiangu Wan

图2 对照指纹图谱(a)和对照品HPLC色谱图(b. 松果菊苷; c. 毛蕊花糖苷; d. 柚皮苷; e. 淫羊藿苷; f. 淫羊藿次苷Ⅱ)Fig. 2 Reference fingerprints(a) and HPLC chromatograms of reference standards(b. Echinacoside; c. Acteoside; d. Naringin;e. Icariin; f. Icariside Ⅱ)
对共有峰进行结构鉴定,共指认了13 种成分。通过对比指纹图谱(图2 谱线a)与对照品保留时间(图2 谱线b-f)指认出其中5个峰: 峰5为松果菊苷; 峰7 为毛蕊花糖苷; 峰8 为柚皮苷; 峰12为淫羊藿苷; 峰19 为淫羊藿次苷Ⅱ。通过串联质谱共有峰分析指认出另外8 种: 峰1 为3,5-二甲氧基-4-羟基苯基β-D-葡萄糖苷; 峰2为新北美圣草苷; 峰3为双藿苷A; 峰9为柚皮素; 峰10为3,6-二芥子酰基蔗糖;峰11 为朝藿定B; 峰13 为鼠李糖基淫羊藿次苷Ⅱ;峰17 为淫羊藿次苷Ⅰ。这些化合物的串联质谱信息如表1 所示,以峰19 的质谱裂解途径为例,简要说明它们的质谱指认过程。峰19 在负离子模式下观察到准分子离子峰m/z513.1760,其主要发生糖基和常见中性小分子的裂解,碎片离子集中在m/z367.1164、352.0912 和324.0966 处。m/z367.1164 的离子对应[M-H-Rha]-,是由一分子鼠李糖基的丢失产生。m/z352.0912 和m/z324.0966 的离子为中性分子的丢失,分别对应[M-H-Rha-CH3]-和[M-H-Rha-CH3-CO]-。通过在质谱中与对照品的保留时间及裂解途径进行比对,峰19被鉴定为淫羊藿次苷Ⅱ,其可能的裂解途径如图3所示。

表1 13种共有峰的质谱鉴定Table 1 Identification of 13 common peaks by mass spectrometry
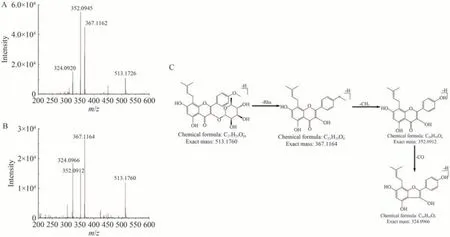
图3 对照品(A)和样品中(B)淫羊藿次苷Ⅱ的二级质谱图及其可能的裂解途径(C)Fig.3 MS/MS spectrum of icariside Ⅱ in reference standard (A) and sample (B), and its possible cleavage pathway (C)
2.3 共有峰归属
对制备的藤黄健骨丸7味药材阴性样品溶液进样测定并记录色谱图,利用“中药色谱指纹图谱相似度评价系统(2012版本)”对色谱图进行分析,结果见图4。比对共有峰发现峰1来源于药材鸡血藤G,峰2、8和9来源于药材骨碎补D,峰3、6、11、12、13、14、15、16、17和19来源于药材淫羊藿F,峰4、20来源于药材熟地黄B,峰5、7来源于药材肉苁蓉E,峰10来源于药材莱菔子H,峰18来源于药材鹿衔草C。
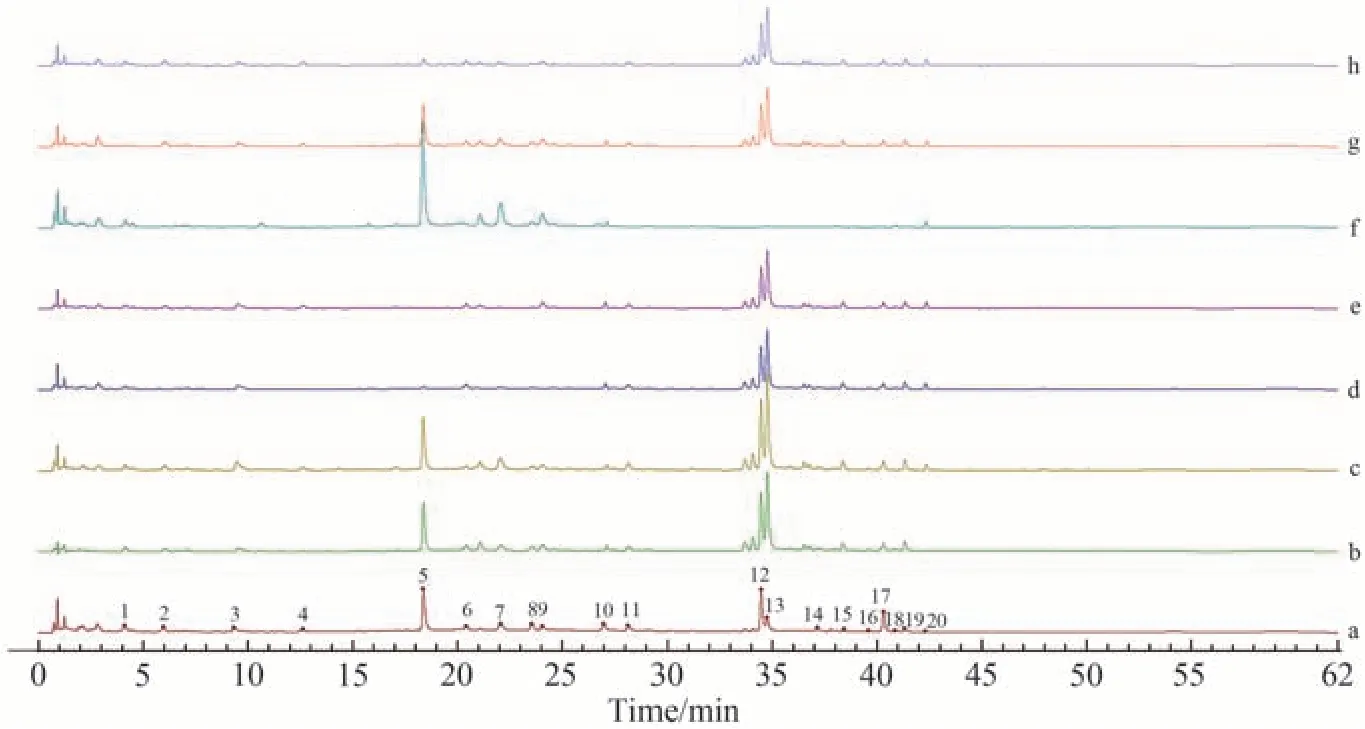
图4 指纹图谱阴性对照Fig.4 Negative controls of fingerprints
2.4 共有峰相对保留时间和相对峰面积计算
设置参照峰(5 号色谱峰)的保留时间和峰面积为1.00,分别计算10 批藤黄健骨丸指纹图谱中其他19 个共有峰相对参照峰的相对保留时间和相对峰面积,结果见辅助材料表S7 和S8。表中结果可以看出,10 批藤黄健骨丸指纹图谱中各共有峰相对保留时间的RSD 值均不超过0.33%,相对峰面积的RSD值均小于4.62%,表明各批次样品的共有峰出峰时间和峰面积均比较稳定,提示它们所代表的各成分含量差异较小,样品质量稳定、可靠。
2.5 指纹图谱相似度评价
取藤黄健骨丸10 批中间体和10 批成品的供试品溶液进行色谱分析并记录色谱图,导入“中药色谱指纹图谱相似度评价系统(2012 版本)”选择X1 作为参照图谱,采用中位数法,时间窗宽度设置为0.1,再进行多点校正以及Mark 峰的匹配,建立10 批成品的指纹图谱,并自动生成对照指纹图谱R。再以生成的对照指纹图谱R为参照图谱,对10批中间体色谱图同法建立中间体-成品指纹图谱。分别计算它们的相似度,结果见表2和3。10批藤黄健骨丸(成品)的指纹图谱相似度均在0.95以上,表明各批样品之间质量均一、稳定,制剂工艺稳定。中间体-成品指纹图谱的相似度在0.954~1.000范围内,说明藤黄健骨丸由中间体加蜜制备成品丸剂的工艺过程中,化学成分的损失较少,主要成分均在中间体及成品中体现,表明中间体与成品间相关性较好,该制剂工艺稳定。
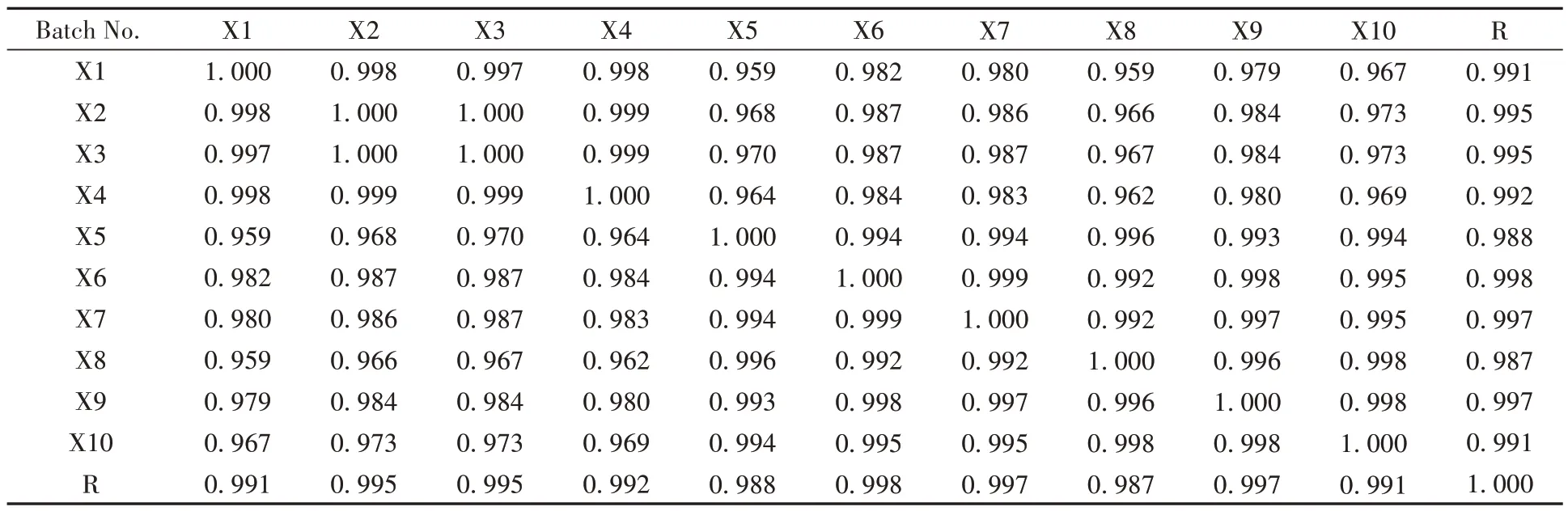
表2 10批成品指纹图谱相似度计算结果Table 2 Fingerprint similarity calculation results of 10 batches finished products
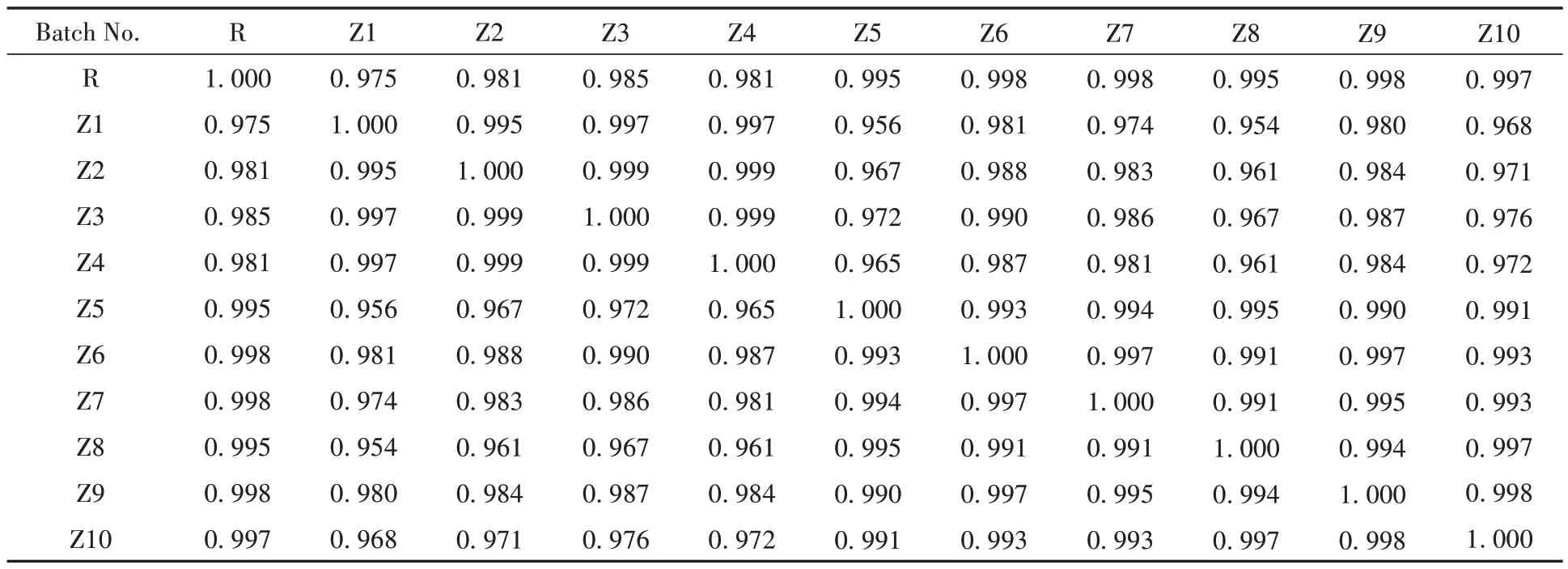
表3 中间体-成品指纹图谱相似度计算结果Table 3 Calculated similarity results of intermediate-control fingerprint
3 结 论
建立了藤黄健骨丸的HPLC指纹图谱,确定了20个共有峰,并参照阴性样品对峰进行了归属。通过对照品保留时间结合串联质谱分析对13 种成分(3,5-二甲氧基-4-羟基苯基β-D-葡萄糖苷、新北美圣草苷、双藿苷A、松果菊苷、毛蕊花糖苷、柚皮苷、柚皮素、3,6-二芥子酰基蔗糖、朝藿定B、淫羊藿苷、鼠李糖基淫羊藿次苷Ⅱ、淫羊藿次苷Ⅰ和淫羊藿次苷Ⅱ)的色谱峰进行了指认,并推断了淫羊藿次苷Ⅱ的质谱裂解途径。计算了10批藤黄健骨丸指纹图谱共有峰的相对保留时间和相对峰面积,结果表明各批次样品共有峰相对保留时间和相对峰面积的差值均较小,所对应的各成分含量保持稳定。对建立的指纹图谱进行了相似度评价,发现10批成品的相似度较好,均在0.95以上,表明该制剂质量稳定,所建立的指纹图谱能对其质量进行整体控制,且藤黄健骨丸中间体与成品之间也具有良好的相关性。综上,本研究建立的指纹图谱可对藤黄健骨丸的内在质量进行整体性评价,为其质量标准提升提供科学依据。
辅助材料(Supporting Information)[指纹图谱相关数据表格]可以从本刊网站(http://yyhx.ciac.jl.cn/)下载。
猜你喜欢
江西中医药(2022年8期)2022-08-22 02:01:26
中国药学药品知识仓库(2022年10期)2022-05-29 02:59:32
汕头大学学报(自然科学版)(2020年4期)2020-12-14 07:04:58
临床医药文献杂志(电子版)(2020年23期)2020-02-28 03:59:52
中国中医药现代远程教育(2018年22期)2018-02-09 02:12:04
湖南林业科技(2017年6期)2018-01-30 03:48:06
吉林大学学报(医学版)(2015年3期)2015-12-17 07:47:42
陕西中医(2015年11期)2015-03-22 04:29:17
安徽中医药大学学报(2014年2期)2014-06-19 13:22:16
长江大学学报(自科版)(2014年23期)2014-03-27 10:51:57
