
JAK/STAT通路介导椎间盘髓核细胞凋亡的研究进展
2024-04-12 02:29:12姬广林
赣南医学院学报 2024年2期
易 军,姬广林
(1. 赣南医科大学第一临床医学院;2. 赣南医科大学第一附属医院骨科,江西 赣州 341000)
作为一种与年龄相关的退行性疾病,椎间盘退变(Intervertebral disc degeneration,IDD)是临床上腰背部疼痛的主要原因之一。一项流行病学研究显示[1],50 岁以上的人群中,约有90%患有IDD。IDD严重影响患者的生活质量,甚至会导致劳动力的丧失,给患者和社会造成沉重的经济负担和精神压力[2]。目前IDD 治疗方法有较大的局限性,其中药物治疗只能稍微缓解患者的疼痛症状,而手术治疗会加速相邻节段的椎间盘发生退变。IDD 可由多种因素诱发,如衰老、遗传因素、机械负荷、肥胖和烟草暴露等。不同信号转导通路在分子、细胞和组织层面参与IDD 病理机制的调控。研究表明Janus 激酶(Janus kinase,JAK)/信号转导子及转录激活因子(Signal transducer and activator of transcriptor,STAT)信号途径在维持椎间盘的正常生理结构和发挥生理功能方面起着重要作用[3]。然而,目前对JAK/STAT信号通路在IDD 发病机制中的作用尚不明确,本文主要就JAK/STAT信号通路的构成、与IDD髓核细胞凋亡的联系以及其作为IDD潜在治疗靶点等方面的研究进展进行综述,以期为IDD的治疗提供参考。
1 椎间盘结构与椎间盘退变
椎间盘(Intervertebral disc, IVD)位于脊柱各椎体之间,牢固地连接上下相邻的椎体,具有缓冲压力和维持脊柱生理活动等作用。IVD 主要由3 个部分组成:髓核、纤维环和软骨终板(Cartilage endplate,CEP)。作为一种无血管的结缔组织,IVD 营养的供给主要依赖于CEP,同时IVD 需要时刻缓冲椎体运动所传递的机械压力,因此IVD 易于损伤并产生退变。在各种病理因素的作用下,IVD 的纤维环结构被破坏,髓核组织结构和功能发生改变,包括细胞外基质(Extracellular matrix,ECM)的进行性降解、细胞衰老速度的加快、髓核细胞凋亡和组织生物力学功能的障碍等,进一步诱导椎间盘代谢失衡,椎间盘高度下降,髓核含水量不断减少,最终导致IDD的发生。
2 椎间盘退变与髓核细胞凋亡
IDD 发病机制复杂多样,至今仍未完全阐明。正常健康IVD的功能主要受组织结构的影响。在衰老、外伤、异常机械负荷、肥胖和炎症等各种病理因素的刺激下,IVD组织内的胶原蛋白和蛋白多糖含量降低,水合能力逐渐下降,IVD 发生进行性退变。在细胞水平上,各种病理因素诱导IVD 细胞尤其是髓核细胞发生代谢紊乱和凋亡,这些病理改变可以反作用于IVD,从而进一步促进IDD 的发生发展。例如,病理因素导致椎间盘细胞自分泌的肿瘤坏死因子α、白细胞介素(Interleukin, IL)等炎症细胞因子水平升高,这些细胞因子通过降低退变椎间盘中的髓核细胞蛋白多糖及胶原纤维的分泌[4]、升高细胞的凋亡率,最终加速椎间盘退变过程[5]。此外,许多与IDD相关的临床前期试验和动物模型的研究也证明了退变IVD 中细胞的凋亡率普遍升高[6-7]。髓核细胞的凋亡和坏死不仅会减少功能细胞的数量,还会释放炎症介质,进一步恶化IVD内的微环境,诱导更多的细胞发生凋亡,最终形成IVD 退化的恶性循环。作为IDD 的病理表现之一,髓核组织纤维化程度增加的频率与髓核细胞数量减少的程度具有一定的相关性。因此,髓核细胞凋亡是IDD 发生发展的重要病理因素之一。
3 JAK/STAT信号通路
有研究[8-10]表明,核因子-κB信号通路、c-Jun 氨基末端激酶信号通路和磷脂酰肌醇-3-激酶/AKT/哺乳动物雷帕霉素靶蛋白通路等参与IDD 的发生发展。作为炎症细胞因子IL-6 最重要的下游途径之一,在肥胖和骨病的研究中发现了JAK/STAT 通路的身影:瘦素和IL-1 之间的协同作用与STAT1 和STAT3的磷酸化有关[11]。在针刺模型诱发椎间盘退变的实验中也发现了IL-6/JAK/STAT 通路参与了针刺模型诱发椎间盘的变性过程[3]。因此,JAK/STAT 信号通路在IDD 中也起着显著的作用,但目前其在IDD中的具体作用机制尚未完全阐明。
JAK/STAT 信号通路主要由酪氨酸激酶相关受体、酪氨酸激酶JAK 以及转录因子STAT 3个部分构成。作为核-膜信号转导途径,JAK/STAT 信号通路虽然构成组分简单,但通过与多种信号分子相互作用,进而参与重要生理活动的调控。在JAK/STAT信号传导过程中,首先不同的细胞因子与其靶细胞表面受体结合后使受体分子二聚化,然后促使JAK磷酸化,接着受体上酪氨酸磷酸化形成的对接位点在SH2 结构域的促进作用下启动招募STAT 蛋白,进而诱导STAT 的羟基酪氨酸位点磷酸化,STAT 活化后从受体复合物中分离并形成二聚体,最后STAT转移入核,通过与特定的DNA 片段结合后发挥其基因调节转录功能[12]。STAT 的活化迅速,活化后的STAT 二聚体进入细胞核中起作用后迅速发生去磷酸化失活,而后再次被转运至胞浆中等待下一次信号转导效应(图1)。
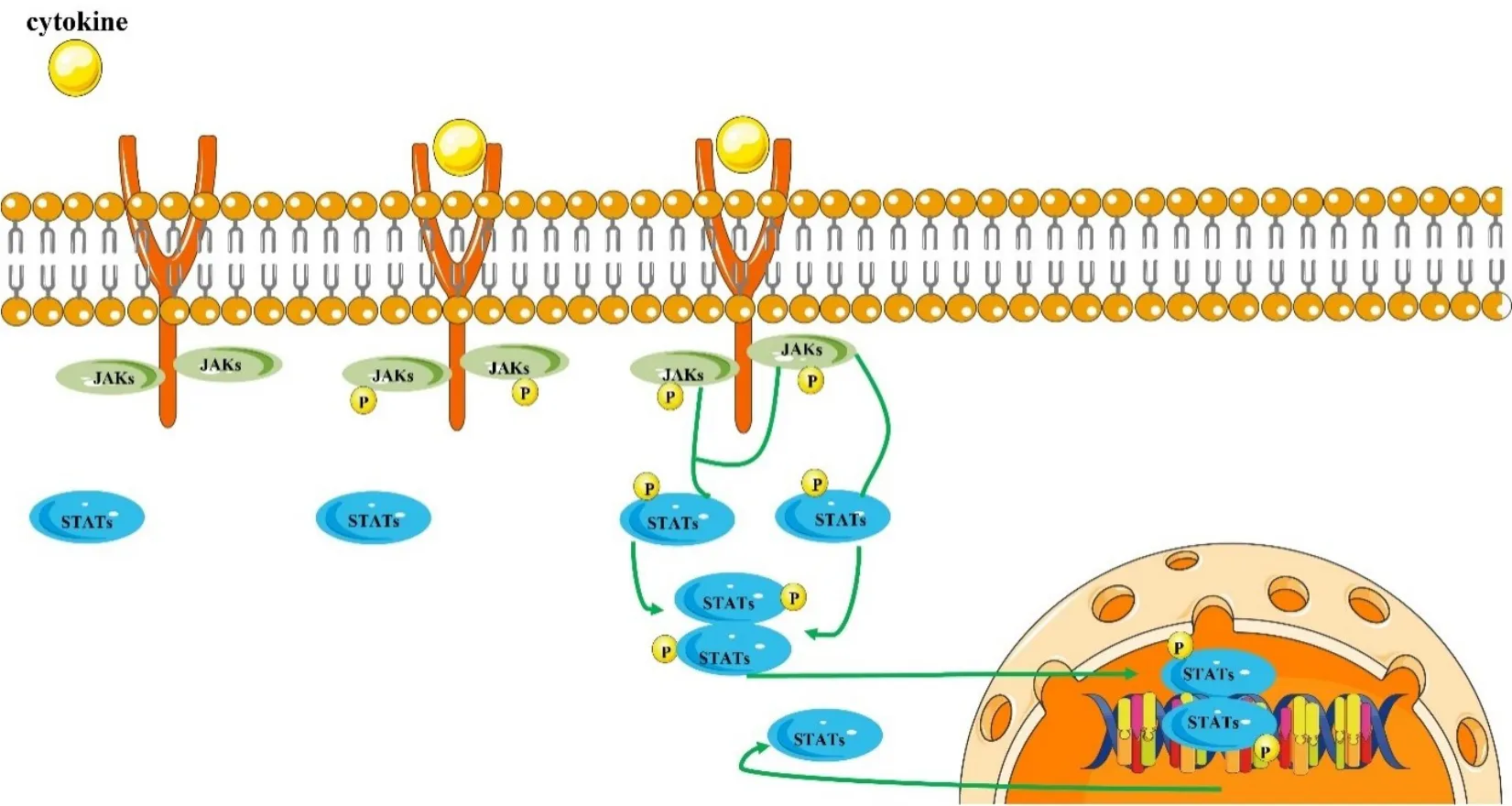
图1 JAK/STAT信号通路示意图
3.1 酪氨酸激酶相关受体酪氨酸激酶相关受体是与细胞因子特异性结合,诱导JAKs 和STATs 活化,进而启动相应生理活动的一种细胞膜型受体[13]。在正常情况下,酪氨酸激酶相关受体并不具有激酶活性。酪氨酸激酶相关受体由含有配体结合位点的细胞外结构域、单次跨膜的疏水α螺旋区、含有蛋白酪氨酸激酶(Protein tyrosine kinases,PTK)活性的细胞内结构域构成。细胞外侧与配体结合,从而接受外部信息,与之相连的是一段跨膜结构,细胞的内侧为酪氨酸激酶活性区域,能促进自身酪氨酸残基的磷酸化以增强该酶活性,再催化细胞内的各种底物蛋白磷酸化,激活胞内蛋白激酶,从而进行细胞信号的传导[14]。
3.2 JAKJAK 属于细胞质中的一种非受体型酪氨酸激酶,JAK 家族包含4 个成员,即JAK1、JAK2、JAK3 和TYK2。JAK3 主要在骨髓、淋巴系统以及内皮细胞和血管平滑肌细胞中表达,其他成员在所有组织中广泛表达[15]。JAK 分子内约含有1 000 个氨基酸残基,其内部有7 个同源域(JH1-JH7),构成JAK 分子内的4个不同的结构域:N 端的FERM 结构域、SH2 结构域、假激酶结构域和激酶结构域。4 个结构域整体相似,第1个结构域是JH1,存在于C端,此结构域也被称为激酶结构域,是4 种JAK 中都含有的催化结构域,主要发挥激酶的酶促活性能力。第2 个结构域JH2 是假激酶结构域,存在于N 端,其酪氨酸激酶活性不足,无催化能力,但JH2对激酶活性的调节作用重大。JAK 还包括与Src-homology-2(SH2)结构域具有同源性的2 个结构域JH3、JH4。JAKs的第4个区域是FERM 结构域,存在于N 端,在JAKs 与细胞因子受体结合时发挥作用[16]。一种细胞因子可以激活多种胞内的JAKs,或不同的细胞因子可以同时激活相同的JAKs 发挥生物学效应。具体的激活机制取决于细胞的类型和细胞所处的微环境[17]。也正是由于JAKs结构的特殊性,其在启动对多种细胞因子受体家族的核-膜信号转导反应中起着关键作用,包含白介素、促红细胞生成素、催乳素等。
3.3 STATSTAT 是JAK 激酶的底物和下游信号分子,它具有进行细胞因子信号转导以及促进特定基因的转录这两个重要的生物学功能。STAT 最初以非活化的形式存在于细胞质中,被磷酸化的JAK激酶活化后STAT 进入细胞核,与核内基因组结合,上调相关基因的表达。但无论是活化形式的STAT还是非活化形式的STAT 均以二聚体的形式存在,每个单体有 STAT1、STAT2、STAT3、STAT4、STAT5a、STAT5b 和STAT6 共7 种亚型。STAT 蛋白从N 端到C 端主要有氨基末端结构域、卷曲螺旋结构域、DNA 结合区、连接结构域、SH2 结构域(Src homology 2 domain, SH2)及C 端的转录激活结构域(Tranional activation domains, TAD)。不同结构域调控STAT 的不同功能,其中N 端结构对STAT 发生磷酸化以及促进二聚体的形成有重要的作用。C 端转录激活区通过丝氨酸磷酸化募集转录激活因子,极大增强了STAT 与DNA 结合后的转录活性。DNA结合区是STAT-DNA 复合物的形成场所。STAT 的SH2 结构域在序列上高度保守,其功能主要是识别细胞因子受体的磷酸酪氨酸序列,与活化的JAK 配合,驱动STAT 的SH2 结构域与磷酸化后的另一个STAT单体的尾部相互作用,形成同源二聚体或异源二聚体[18]。TAD 在不同的STAT 成员之间有很大的差异,这使得不同的STAT 可以激活不同类型基因和通路的转录表达,呈现其成员间功能上的不同,如STAT1 参与抑制细胞生长、调节细胞分化、促进细胞凋亡、调节免疫等[19-20];STAT2 能够参与细胞的抗病毒作用、免疫调节、抗凋亡以及对肿瘤发生发展的调控等[21];STAT3 主要参与免疫反应细胞的负调控,调控细胞的生长、分化、凋亡以及肿瘤发生、转移等[22]。不同的细胞因子倾向于激活特定的STAT,但细胞因子之间的相互作用也会在一定程度上影响同一STAT的作用[23]。这使得JAK/STAT信号通路在各种代谢活动的调节中均扮演着重要角色。
4 JAK/STAT信号通路与IDD髓核细胞凋亡的联系
JAK/STAT 通路在IDD 的发病机制中扮演着至关重要的作用。研究证实,IL-6 等细胞因子可以通过激活JAK/STAT信号通路诱导细胞凋亡,且IDD的动物模型中也可以同时观察到JAK/STAT 信号通路的相关靶蛋白表达上调和髓核细胞凋亡增加[24]。此外,抑制JAK/STAT通路可以降低IDD中髓核细胞的凋亡率[25]。因此,JAK/STAT信号通路可能通过介导髓核细胞凋亡而影响IDD 的进程,但具体机制尚未明确。
4.1 IDD 发展过程中JAK/STAT 信号通路与ECM 降解、髓核细胞凋亡和炎症之间的作用
ECM 降解、细胞凋亡和炎症被称为椎间盘退行性疾病的三大标志,它们三者之间相互联系、相互影响[26]。促炎细胞因子通过上调ECM 降解酶的表达并干扰ECM 的组织学结构,进而诱导ECM 代谢失衡[27]。ECM 本身的降解会产生过量活性氧,导致细胞代谢调节能力下降,进一步刺激髓核细胞的炎症反应[28]。此外,IVD组织中较高的细胞凋亡率及衰老率和较低的ECM 合成能力与炎症有关,炎症在诱导IDD 中的髓核细胞凋亡起重要作用。促炎因子,如IL-1β已被证明可以诱导髓核细胞凋亡,IVD中较高水平的IL-6与IDD进展相关[29]。椎间盘突出症患者在硬膜外给予抗IL-6 受体单克隆抗体(Tocilizumab)后,可缓解腰痛和根性腿痛、麻木等症状[30]。研究表明,JAK/STAT信号通路正是介导炎症与IDD 关联的重要中间介质。WU C H 等[25]在一项研究中发现白藜芦醇可抑制人髓核细胞的细胞凋亡和细胞周期停滞,其机制可能与抑制JAK/STAT3磷酸化和IL-6 产生减少有关。MIAO D 等[24]研究表明,通过瘦素激活JAK2/STAT3 途径促进大鼠髓核细胞的分解代谢,结果显示髓核细胞中Ⅱ型胶原的表达量相对降低。而Ⅱ型胶原表达减少可促进髓核细胞的退化和凋亡[31]。同时,研究[32]发现JAK1/STAT3信号传导途径可能也参与了髓核细胞凋亡的调节过程。CHEN B 等[33]在实验中发现IL-21 可通过STAT 信号通路刺激炎症因子TNF-α 间接促进IDD 的进展,其中以STAT3 的表达最强。DU X X等[34]在研究中发现miR-223 可通过JAK2/STAT1 信号途径调控髓核细胞凋亡,进而促进椎间盘ECM 的降解和IDD 的发生。总之,JAK/STAT 通路通过干预ECM 降解与炎症反应调节IDD 中的髓核细胞凋亡,但具体作用机制仍有待进一步研究。
4.2 IDD发展过程中JAK/STAT信号通路与内质网应激、髓核细胞凋亡之间的关系内质网是体内蛋白质加工折叠的工厂。一旦内质网受到病理因素的严重刺激就会诱发内质网应激(Endo plasmicreticulum stress,ERS)从而影响蛋白质的折叠。IVD 作为重要的器官,需要合成和加工大量的胶原蛋白和蛋白聚糖以维持功能,因此IVD 的内质网更易于发生ERS。研究表明,机械负荷过大、营养缺乏、代谢紊乱、炎症等多种病理因素可诱发ERS的慢性激活并诱导椎间盘ECM 降解和髓核细胞凋亡等从而进一步促进IDD的进展[35]。目前尚无直接证据表明IDD 发病进程中ERS 与JAK/STAT 信号的联系。然而,有研究表明人类丙型肝炎病毒通过激活ERS 而上调STAT3 的蛋白表达[36],同时研究发现,在原代星形胶质细胞中PERK/JAK1/STAT3通路被激活以响应ERS 诱发的炎症[37]。总体而言,这些发现表明STAT3 在ERS 中的影响,但STAT3 是如何被激活以响应ERS,特别是在IDD 髓核细胞凋亡中的机制需要进一步研究(图2)。
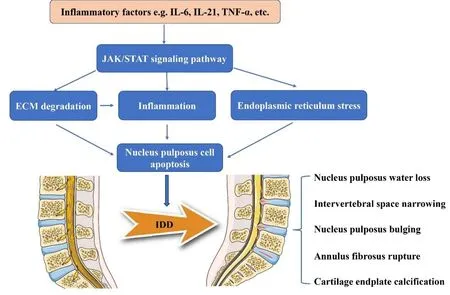
图2 JAK/STAT信号通路调控IDD髓核细胞凋亡示意图
5 JAK/STAT信号通路作为IDD的潜在治疗靶点
目前以JAK/STAT 信号通路作为靶点开发相关药物治疗IDD 的相关研究相对较少。抑制JAK/STAT 信号通路的抑制剂主要包括细胞因子信号转导负调节因子(SOCS 蛋白)、JAK 抑制剂、STAT 蛋白抑制剂、蛋白酪氨酸磷酸酶(Protein tyrosine phosphatases, PTPs)等[13]。SOCS 家族包含有细胞因子诱导的含sh2 蛋白(CIS)及SOCS1~7 共8 种类型[38]。其中SOCS1和SOCS3可抑制JAK1、JAK2和TYK2激酶活性,但不能抑制JAK3 的活性[39]。有趣的是,当STATs 激活后可以促进SOCS 蛋白的表达,而SOCS蛋白在与磷酸化的JAK和JAK受体结合后反而会抑制JAK/STAT 信号通路的信号传导[40]。研究发现[32]在腰椎间盘突出患者中有一定量的SCOS3 蛋白表达,因此SOCS 基因或许可以作为治疗IDD 的靶基因,为开发治疗IDD的相关药物提供新的方向。
当前市场上及临床试验中常用的JAK 抑制剂有鲁索利替尼(Ruxolitinib)、托法替尼(Tofacitinib)、AG490、巴瑞替尼(Baricitinib)、菲卓替尼(Fedratinib)等[41]。不同的抑制剂对JAKs 具有不同的选择性及作用强度,如鲁索利替尼是一种对JAK1 和JAK2 的选择性强效抑制剂,托法替尼是JAK1 和JAK3 的选择性强效抑制剂。JAK抑制剂作为新的小分子靶向药物,已成功应用于类风湿性关节炎等疾病的治疗[42]。随着对JAK 靶点科学研究的逐步深入,JAK抑制剂在自身免疫性疾病的应用中也取得了较好的治疗效果[43]。鉴于JAK 抑制剂的应用前景,探究JAK 抑制剂治疗IDD 的分子机制已成为相关研究的热点内容,但遗憾的是,目前尚无有效的药物被批准用于IDD的临床治疗。未来JAK抑制剂有望成为治疗IDD潜在的靶点,有待进一步深入研究。
目前常用的STAT 抑制剂主要有Stattic、AG490、氟达拉滨(Fludarabine)、青蒿琥酯等,但STAT 抑制剂目前的应用焦点是抗肿瘤治疗。尽管已引入了多种STAT 抑制剂,但由于其选择性、生物利用度、相似的同源性及在生物体内功效不一等问题,目前尚未成功开发适用于临床的STAT 抑制剂[44]。因此,能否将STAT抑制剂应用于IDD仍需进一步深入研究。
PTPs 家族与PTK 共同维持酪氨酸蛋白磷酸化的平衡,参与细胞的信号转导,调节细胞的生长、分化、代谢、基因转录和免疫应答等。PTPs 通过与JAK、STAT 或酪氨酸激酶相关受体相互作用来抑制JAK/STAT通路,主要方式有:⑴使STAT二聚体去磷酸化;⑵与酪氨酸激酶相关受体相互作用,使相关的JAK 去磷酸化;⑶在CD45(一种通过去磷酸化过程传导细胞信号的跨膜PTP)的作用下,抑制JAK 的磷酸化[45]。适当水平的酪氨酸磷酸化对于维持多种细胞生理活动至关重要,但由于突变、调节异常或PTP表达改变而发生的异常酪氨酸磷酸化与许多人类疾病和紊乱有关[46]。正因如此,很少有研究将其应用于以JAK/STAT作为靶点治疗IDD。
6 小结与展望
在IDD 的发展过程中,髓核细胞扮演着至关重要的角色,炎症、髓核细胞凋亡和细胞外基质的降解都能减少髓核细胞的数量,从而诱发IDD。髓核细胞的正常功能的维持取决于ECM 中Ⅱ型胶原和蛋白聚糖之间的合成与分解代谢平衡,此种平衡又依赖于机体内复杂的代谢通路中的信号分子对髓核细胞微环境的调节。在IDD 中,髓核细胞的合成代谢弱于分解代谢,这促进了细胞外基质的降解和髓核细胞凋亡。在细胞凋亡、ERS 和炎症等多种重要生理过程中JAK/STAT 信号通路均表现出重要的作用,但目前的研究表明JAK/STAT 通路主要受炎性细胞因子的激活从而调控髓核细胞的凋亡来影响IDD 的发病,其是如何通过介导ERS 及自噬来调控髓核细胞凋亡进而影响IDD 需进一步深入研究。虽然目前临床试验中大多数针对JAK/STAT 信号通路的药物对IDD 的有效性以及确切的机制尚不清楚,但JAK/STAT通路仍有望成为IDD重要的治疗靶点。进一步探究JAK/STAT 通路有助于阐明IDD 的具体发病机制,这对未来IDD 的防治与提高IDD 患者生活质量具有重大的临床意义。
猜你喜欢
天津医科大学学报(2021年3期)2021-07-21 09:03:46
世界科学技术-中医药现代化(2021年12期)2021-04-19 12:31:40
广州大学学报(自然科学版)(2019年1期)2019-05-07 01:33:26
天然产物研究与开发(2018年1期)2018-02-02 07:21:22
中成药(2018年1期)2018-02-02 07:19:57
中国塑料(2016年7期)2016-04-16 05:25:52
天津科技大学学报(2016年1期)2016-02-28 16:59:45
湖北师范大学学报(自然科学版)(2015年2期)2016-01-10 08:41:53
中国医药生物技术(2015年4期)2015-12-26 08:26:36
现代检验医学杂志(2015年2期)2015-02-06 02:01:01
