
石墨烯/钛界面合金元素偏聚与界面反应抑制机理
2024-04-08 10:32陈佳莹熊小倩黄敬涛李明伟来忠红朱景川
中国材料进展 2024年3期
陈佳莹,熊小倩,黄敬涛,曲 囡,李明伟,程 源,来忠红,刘 勇,朱景川
(1. 哈尔滨工业大学材料科学与工程学院,黑龙江 哈尔滨 150001)
(2. 哈尔滨工业大学 特种环境复合材料技术国家级重点实验室,黑龙江 哈尔滨 150001)
(3. 哈尔滨工业大学 分析测试与计算中心,黑龙江 哈尔滨 150001)
1 前 言
钛及其合金由于具有相对较低的密度、良好的力学性能和高耐蚀性,广泛应用于汽车、航空航天、国防以及生物医学等领域[1-4]。但是目前钛合金的应用仍受到许多因素的限制,例如合金强度、模量和耐磨性的不足等[5-9]。为了改善这些问题,最有效的方法就是通过引入第二相去增强钛合金的力学性能,即制造高性能钛基复合材料(titanium matrix composites,TMCs)[10-12]。石墨烯具有较高的强度和弹性模量,与钛及钛合金复合有望开发出具有优异力学性能的TMCs[13]。然而,由于金属钛性质活泼,非常容易与碳发生化学作用[14],产生大量界面反应物,使得制备高性能石墨烯/Ti基复合材料变得困难。因此,石墨烯与Ti基体之间的界面结合强度将对复合材料的整体性能产生显著影响,正确了解界面的微观结构信息与性质对实现复合材料整体性能的控制具有重要的意义[15-17]。
第一性原理可用于深入研究两相界面的结合强度、稳定性、原子结构和电子结构,可对界面性能做出合理预测[17-19]。何小晶等[20]建立了石墨烯吸附Ti原子结构模型,发现Ti原子能稳定吸附于石墨烯六元环正上方,并且缺陷石墨烯可以大大提高石墨烯的吸附特性。Zhang等[21]基于第一性原理提出了石墨烯/金属基复合材料的金属掺杂和空位石墨烯的界面模型,以控制金属-石墨烯之间的界面,进一步强化金属-石墨烯复合材料的力学性能。Yi等[22]采用第一性原理计算了Ti/石墨烯/Ti界面的原子结构,发现Ti和C原子间的结合兼具共价键、离子键特点;随着石墨烯层数增加,Ti/石墨烯/Ti界面更稳定,结合更强。Peng等[23]发现在石墨烯薄片上镀铜可提高石墨烯-Al基复合材料的力学性能。与未镀铜复合材料相比,镀铜3%(质量分数)的石墨烯铝基复合材料的抗拉强度提高了77.5%。然而,目前关于合金元素对石墨烯/Ti基复合材料界面性质及界面反应机理研究较少。因此,深入研究合金元素对石墨烯/Ti基复合材料界面的影响,对优化界面结构从而提高复合材料性能具有重要的理论和实际意义。
本文运用第一性原理方法,建立石墨烯吸附双原子模型以及合金元素掺杂Ti/石墨烯/Ti界面模型,通过研究偏聚能的变化、原子位置的变化、电子的得失转移以及成键情况等,从根本上研究合金元素对Ti/石墨烯/Ti界面的作用机理,根据不同合金元素对Ti/石墨烯/Ti界面偏聚行为和电子结构的影响,筛选出可能抑制界面反应的合金元素,为优化钛合金体系、制备高性能TMCs提供理论指导。
2 计算模型与参数
本文所有计算基于第一性原理中的CASTEP代码[24-26]实现密度泛函理论。所有计算均以平面波为基础,使用超软赝势[24,27,28]。选取Vanderbilt超软赝势和平面波基组展开体系波函数[29],交换关联能量泛函采用广义梯度近似中的佩德乌-伯克-恩泽霍夫泛函形式来描述电子间相互作用[30]。布里渊区k点取样采用Monkhorst-Pack网格,费米能计算采用高斯能带展宽方法。合金元素掺杂Ti/石墨烯/Ti界面模型的截断能为540 eV,k点为8×8×1。计算中的其他相关参数设定如下:自洽场公差等级、布罗伊登-弗莱彻-戈德法布-山诺算法等级均为fine,高斯能带展宽值δ为0.1 eV。
先前研究表明[23],钛的(0001)表面因具有特殊晶体结构和电子布局而极为稳定。此外,Ti(0001)面能与石墨烯的(0001)面形成高度匹配的界面,显著降低错配度和能量成本。这不仅增强了界面稳定性,也为研究合金元素在界面处的行为提供了理想环境。并且金属与石墨烯容易发生界面反应的层数一般为1~2,所以对Ti超胞进行表面切割,切割厚度为3个原子层,保证界面反应的进行。在构建模型时,为保证石墨烯与Ti之间的界面失配度尽可能小,对石墨烯和Ti表面进行了4×4、3×3的扩胞,搭建了Ti(0001)/Gra(0001)/Ti(0001)的“三明治”模型,点阵失配度在5%以内。再将最近层和次近层Ti原子的中间位置替换为合金元素,使界面间距值为2.30 Å,最终建立的Ti/石墨烯/Ti界面掺杂合金元素的界面结构模型如图1所示[22]。
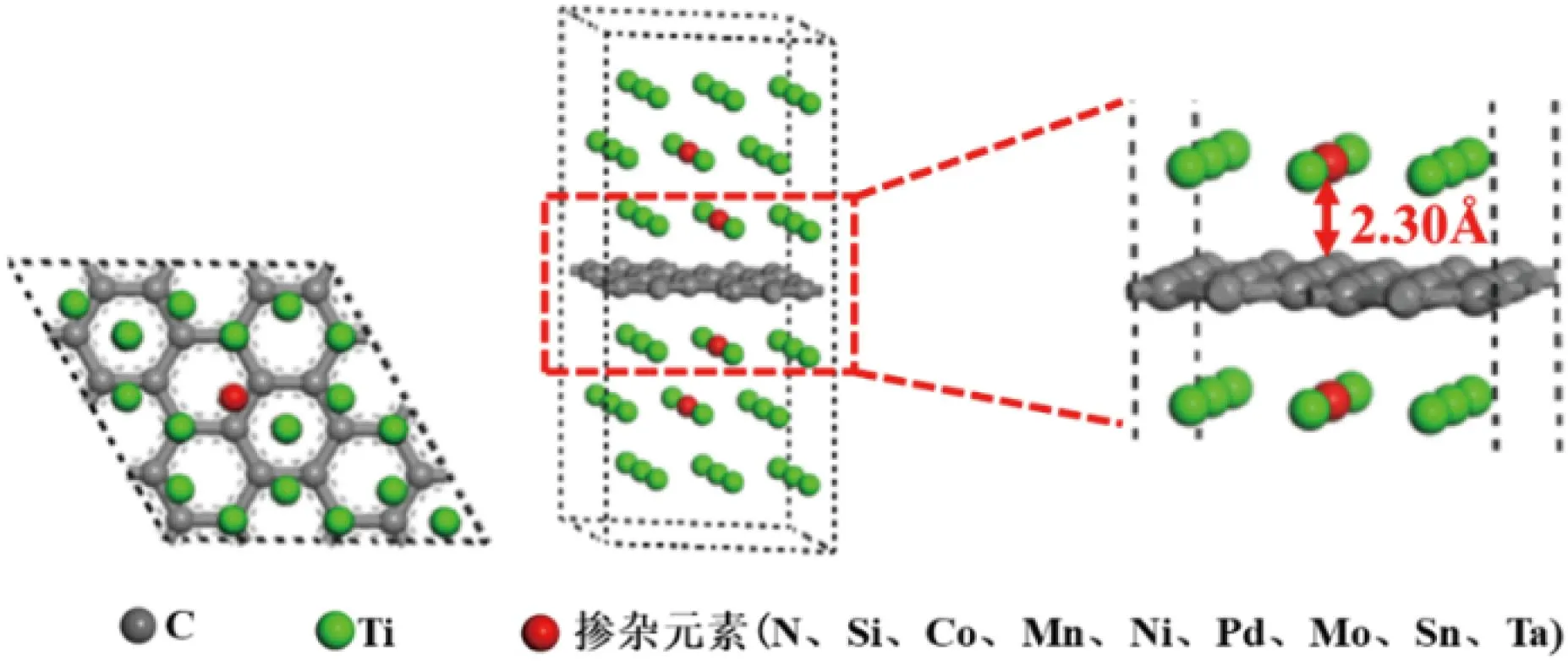
图1 Ti/石墨烯/Ti界面掺杂合金元素模型[22]Fig.1 Model of Ti/graphene/Ti interface doped alloying element[22]
3 结果与讨论
3.1 界面偏聚能
合金元素对界面结合能力和稳定性的影响可以通过界面偏聚能来衡量[23]。在本研究中,计算得到的界面偏聚能揭示了不同合金元素在Ti/石墨烯界面的迁移倾向。界面偏聚能越小说明合金原子越容易向界面偏聚,反之则容易向基体偏聚。可通过计算溶质原子从晶界较远处偏聚到晶界区域的能量变化获得溶质的偏聚能[24],偏聚能ΔEseg的计算公式如下:
(1)
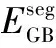
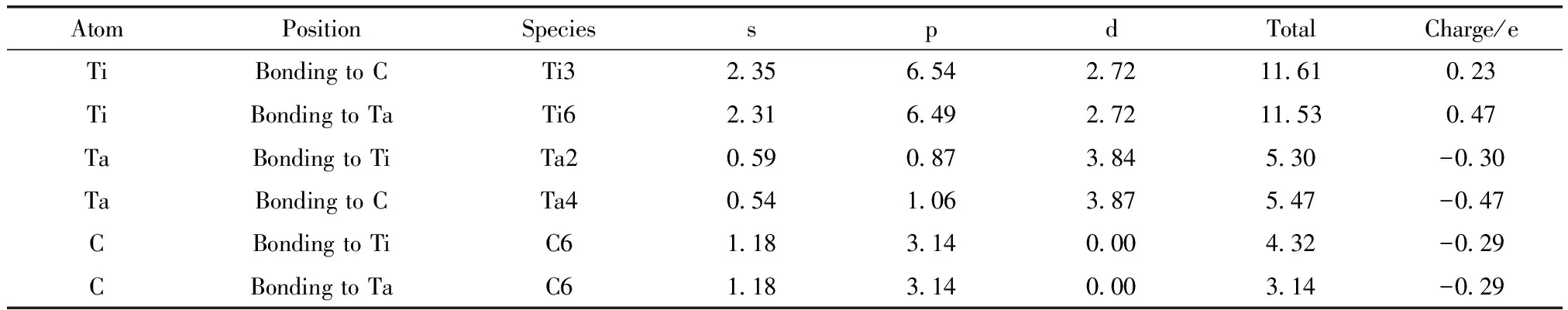
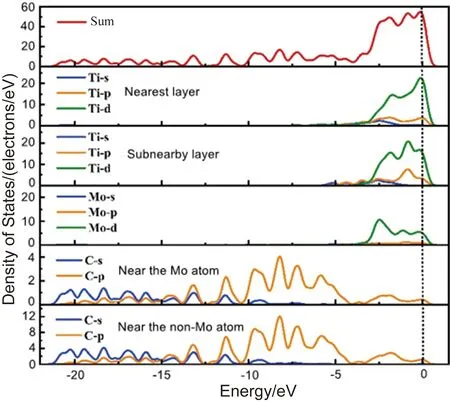
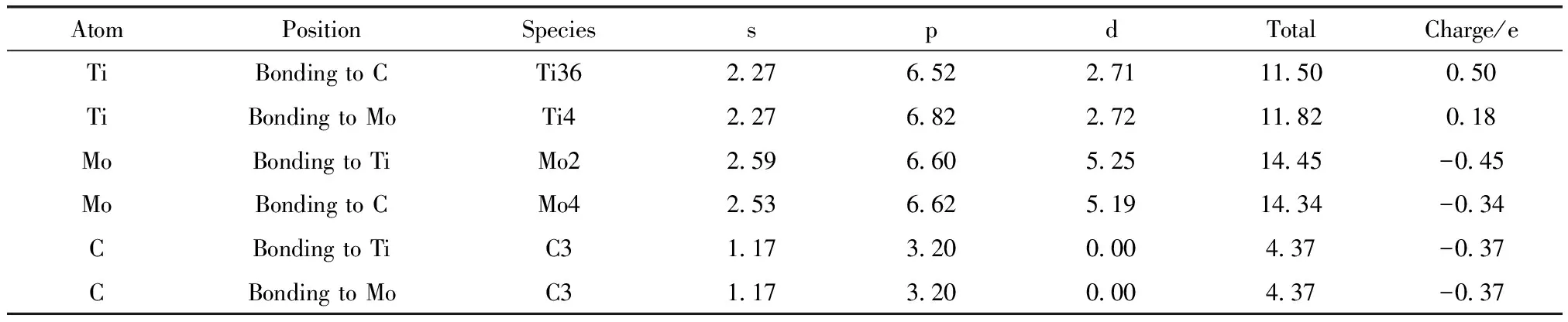
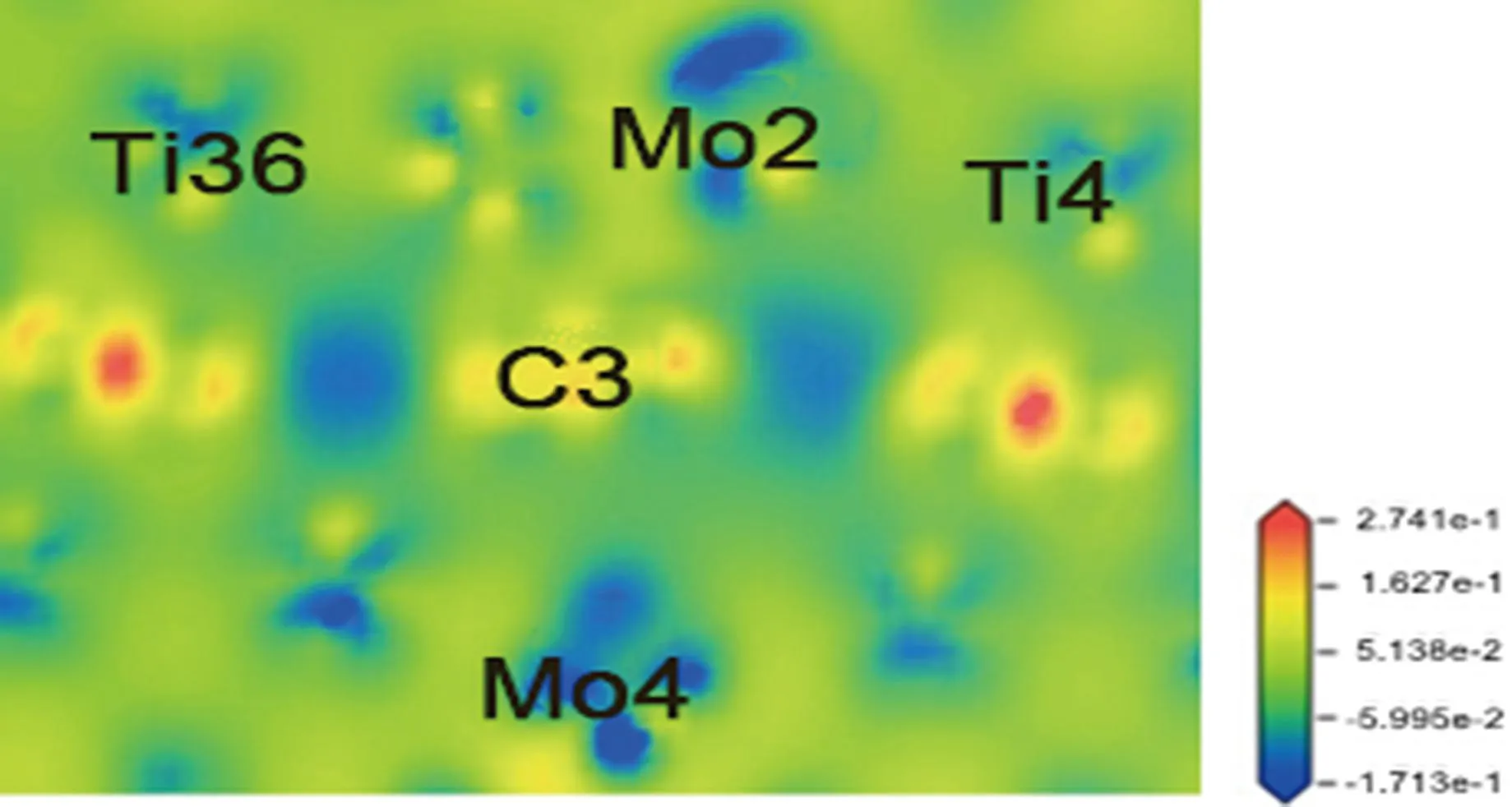
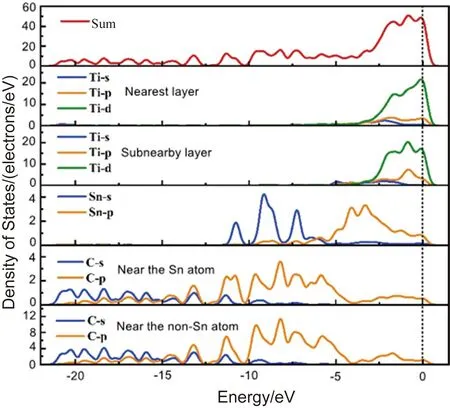
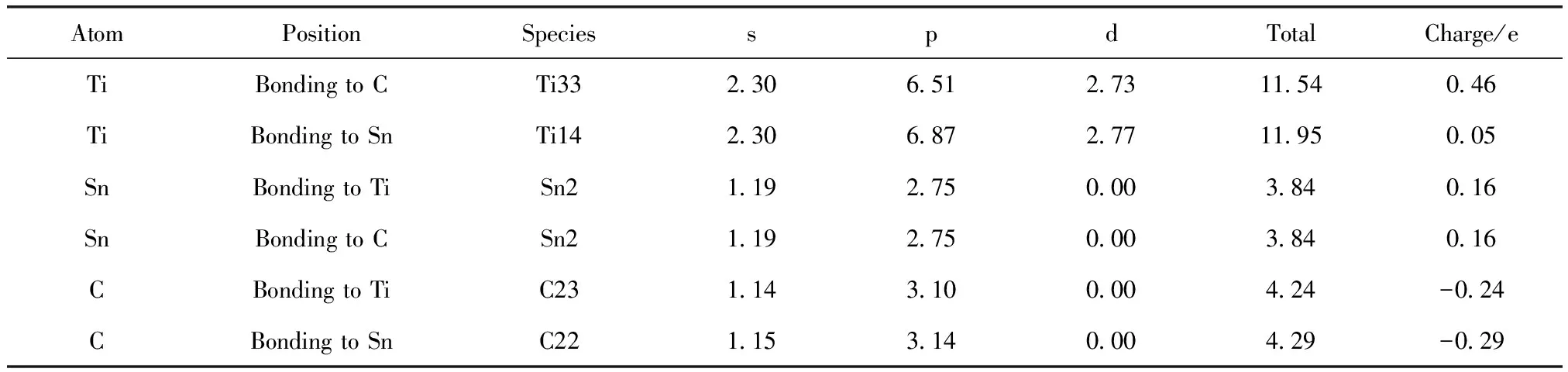
偏聚能数值越负代表溶质原子的偏聚能力越强,界面结合能力越强,越利于提升界面稳定性。计算得到掺杂不同元素的界面偏聚能如图2所示,由图可知,偏聚能的排序为:Ta>Mo>Sn>Pd>Si>Ni>Co>Mn>N,最大值为-16.79 eV,最小值为-38.63 eV。因此,界面结合能力排序为:Ta 图2 Ti/石墨烯/Ti界面掺杂合金元素的界面偏聚能Fig.2 Interface partial polyet energy of Ti/graphene/Ti interface doped alloy element 图3为几何优化后的Ti/石墨烯/Ti界面模型,弛豫后的石墨烯均产生了明显的褶皱,显示了界面层原子与石墨烯强烈的相互作用,并且与未掺杂合金元素的界面模型相比,不同元素掺杂后各个C原子产生位置偏移的程度各不一样,导致石墨烯产生褶皱的程度不同,说明掺杂合金元素在一定程度上改变了Ti与石墨烯之间强烈的相互作用。由图中可以明显看出掺杂Ta原子时,界面处Ti原子层位置偏移较小,几乎没有发生位置波动,界面处的Ta原子也几乎没有产生位置偏移,符合Ta原子界面偏聚能最大的规律。界面层Ta原子与石墨烯层(z轴平均坐标)之间的界面间距为2.180 Å,界面层Ti原子与石墨烯层(z轴平均坐标)之间的最小间距为1.941 Å,最大间距为2.311 Å。当掺杂Mo原子时界面层Ti原子层的位置波动较大,Mo原子与石墨烯层(z轴平均坐标)之间的界面间距变为2.715 Å,界面层Ti原子与石墨烯层(z轴平均坐标)之间的最小间距为2.035 Å,最大间距为2.168 Å。掺杂Sn原子时界面层原子位置偏移也较大,Sn原子与石墨烯层(z轴平均坐标)的界面间距为2.826 Å,界面层Ti原子与石墨烯层(z轴平均坐标)之间的最小间距为2.268 Å,最大间距为2.197 Å。 图3 几何优化后的Ti/石墨烯/Ti界面模型Fig.3 Interface mode of geometrically optimized Ti/graphene/Ti 在Ti/石墨烯/Ti界面引入合金元素,对界面的电子分布产生明显影响,计算的态密度结果如图4所示,对Ti-Ta/石墨烯/Ti-Ta界面不同位置的Ti,Ta,C原子的态密度曲线进行分析。分析得出:最近层和次近层Ti原子的2p轨道、3d轨道与C原子2p轨道、1s轨道杂化,在费米能级附近产生新的共振峰,与纯Ti/石墨烯/Ti界面相比,共振峰发生了红移,这说明Ti原子与C原子之间的相互作用被减弱。Ti原子的2p轨道、3d轨道与Ta原子的2p轨道、3d轨道杂化,从费米能级附近Ti原子与Ta原子存在的共振峰可以看出明显的Ti—Ta键。由Ta原子附近和非Ta原子附近C的分波态密度可以看出,Ta原子和C原子之间存在共振峰,存在C—Ta键。 对Ti-Ta/石墨烯/Ti-Ta界面进行原子轨道布局和不同编号原子的键布局分析,结果如表1和表2所示。分析原子轨道布局可知,与C成键的Ti原子失去电子0.23e,C原子得电子0.29e,Ti原子与C原子有离子键结合的特点,但与未添加合金元素界面相比,C原子得电子数减少,这表明Ta元素的加入削弱了Ti与C之间的结合。Ta和Ti原子与C原子成键时皆得到电子,分别为0.30e和0.47e。结合键布局,界面模型中存在C—Ti键、Ti—Ta键和C—Ta键,键长分别为2.79171,2.82175和2.82995 Å,与态密度分析结果一致,且分别具有离子键、共价键和离子键结合特点。 表1 Ti-Ta/石墨烯/Ti-Ta界面原子轨道布局 表2 Ti-Ta/石墨烯/Ti-Ta界面键布局 对Ti-Ta/石墨烯/Ti-Ta界面进行差分电荷密度计算,结果如图5所示,相比于Ti/石墨烯/Ti界面,Ti3和Ti6原子周围电荷密度均减少,Ta2原子周围电荷密度有增大也有减少,C6原子周围电荷密度增大,说明C原子不仅从Ti原子处获得了电子,也从Ta原子处获得了电子,并且Ta原子也从Ti原子处获得电子,因此在一定程度上减小了Ti原子和C原子之间的电子转移,即减弱了Ti与C之间的结合能力。 图5 Ti-Ta/石墨烯/Ti-Ta界面差分电荷密度图Fig.5 Differential charge density diagram of Ti-Ta/graphene/Ti-Ta interface 为了考察Mo元素对Ti/石墨烯/Ti界面电子结构的影响,计算了Ti-Mo/石墨烯/Ti-Mo界面不同位置的Ti,Mo,C原子的态密度曲线,如图6所示。可以看出与Ti-Ta/石墨烯/Ti-Ta界面相同,最近层和次近层Ti原子的3d轨道与C原子2p轨道杂化,在费米能级附近产生新的共振峰,与纯Ti/石墨烯/Ti界面相比,共振峰发生了红移,这说明Ti原子与C原子之间的相互作用发生了一定的减弱。Ti的2p轨道与Mo的2p轨道产生杂化,在-0.180 eV处产生明显的共振峰,说明存在Ti—Mo键。由Mo原子附近和非Mo原子附近C的分波态密度差异可以看出,Mo原子和C原子之间在费米能级附近存在弱的共振峰,存在C—Mo键。 图6 Ti-Mo/石墨烯/Ti-Mo界面态密度Fig.6 Interface density of Ti-Mo/graphene/Ti-Mo Ti-Mo/石墨烯/Ti-Mo界面原子轨道布局如表3所示,Ti原子的各个轨道均失去电子,与C成键的Ti原子失去电子0.50e,与Mo成键的Ti原子失去电子0.18e。Mo原子各个轨道均得到电子,与Ti成键的Mo原子得到电子0.45e,与C成键的Mo原子得到电子0.34e。C原子各轨道也得到电子,既与Ti又与Mo成键的C原子得到电子0.37e,与纯Ti/石墨烯/Ti界面相比得到的电子数增多,这说明Mo原子也向C原子转移了一部分电子。结合Ti-Mo/石墨烯/Ti-Mo界面键布局(表4)可知,界面模型存在C—Ti键、Ti—Mo键和C—Mo键,与态密度分析一致,分别为共价键和离子键结合特点。 表3 Ti-Mo/石墨烯/Ti-Mo界面原子轨道布局 表4 Ti-Mo/石墨烯/Ti-Mo界面键布局 Ti-Mo/石墨烯/Ti-Mo界面差分电荷密度如图7所示,可以看出,不仅C3原子从Ti36原子处得到电子,Mo2原子也从Ti4原子处得到电子,因此Mo原子的存在减弱了Ti原子与C原子之间的结合。Mo4周围电荷密度有增大也有减小,说明C3和Mo4成离子键结合特点。 图7 Ti-Mo/石墨烯/Ti-Mo界面差分电荷密度图Fig.7 Differential charge density diagram of Ti-Mo/graphene/Ti-Mo interface 计算的Ti-Sn/石墨烯/Ti-Sn界面的态密度如图8所示,Ti原子与C原子产生共振峰的位置红移,Sn原子的加入在一定程度上影响了Ti与C之间的相互作用力,与上述加入Ta和Mo元素分析一致。Ti的3d轨道和Sn的2p发生杂化,产生新的共振峰,表明有Ti—Sn键的存在。Sn原子附近的C和非Sn原子附近的C与Sn产生较弱共振峰,表明C原子和Sn原子成键较少。 图8 Ti-Sn/石墨烯/Ti-Sn界面态密度Fig.8 Interface density of Ti-Sn/graphene/Ti-Sn 计算的Ti-Sn/石墨烯/Ti-Sn界面的原子轨道布局、键布局如表5和表6所示,分析可得与Ti原子和C原子成键的Sn原子失去电子0.16e,并且结合键布局结果可知C—Sn键数目只有1个,与态密度分析一致,均为离子键结合特点。对比Ta原子、Mo原子的原子轨道布局数还可以看出,当C原子与Sn原子成键时,C原子与Ti原子未成键,这表明,Sn元素的加入减弱了Ti原子与C原子之间的结合。 表5 Ti-Sn/石墨烯/Ti-Sn界面原子轨道布局 表6 Ti-Sn/石墨烯/Ti-Sn界面键布局 Ti-Sn/石墨烯/Ti-Sn界面差分电荷密度如图9所示,Sn2周围原子电荷密度变化不大,Sn2原子向C22原子和Ti33原子转移电荷,减少了Ti原子电荷向C原子的转移,结合键布局分析可知,Ti14与C23未成键,Sn元素的加入削弱了Ti与C之间的结合。Ti原子周围电荷密度有增大有减小,说明Ti与C之间不仅有共价键结合的特点,也有离子键结合的特点。 图9 Ti-Sn/石墨烯/Ti-Sn界面差分电荷密度图Fig.9 Differential charge density diagram of Ti-Sn/graphene/Ti-Sn interface 基于密度泛函理论的第一性原理计算了合金元素掺杂Ti/石墨烯Ti界面的界面偏聚能,以此进行了合金元素筛选,并分析了最终筛选出的合金元素对Ti/石墨烯/Ti界面偏聚行为和电子结构的影响,得出结论如下: (1)掺杂不同合金元素的Ti/石墨烯/Ti界面偏聚能大小为:Ta>Mo>Sn>Pd>Si>Ni>Co>Mn>N,偏聚能数值越负代表溶质原子的偏聚能力越强,界面结合能力越强,所以最终筛选出Ta,Mo和Sn这3种可能减弱石墨烯与Ti之间界面结合的合金元素; (2)分析Ta,Mo和Sn对界面偏聚行为和电子结构影响得出,合金元素的加入减弱了界面层原子的偏移程度,减少了Ti与C之间的电荷转移,即减弱了界面结合。 本研究对钛合金体系的优化和高性能钛基复合材料的制备具有重要的理论和实际意义。对航空航天、医疗和汽车行业等领域也具有参考价值,为理解合金元素偏聚行为提供了新视角,并为这些行业中高性能钛基复合材料的设计问题提出了一种解决思路。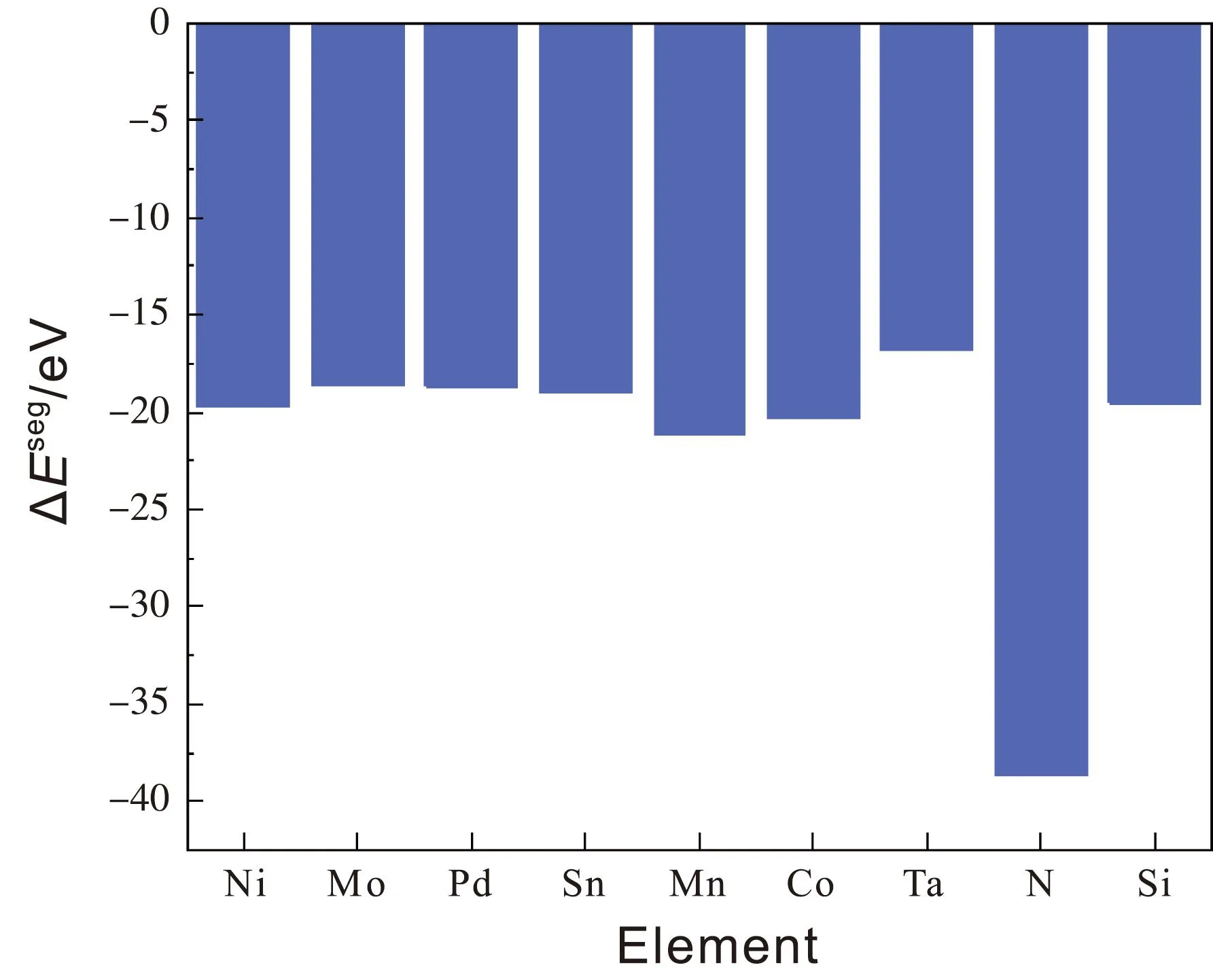
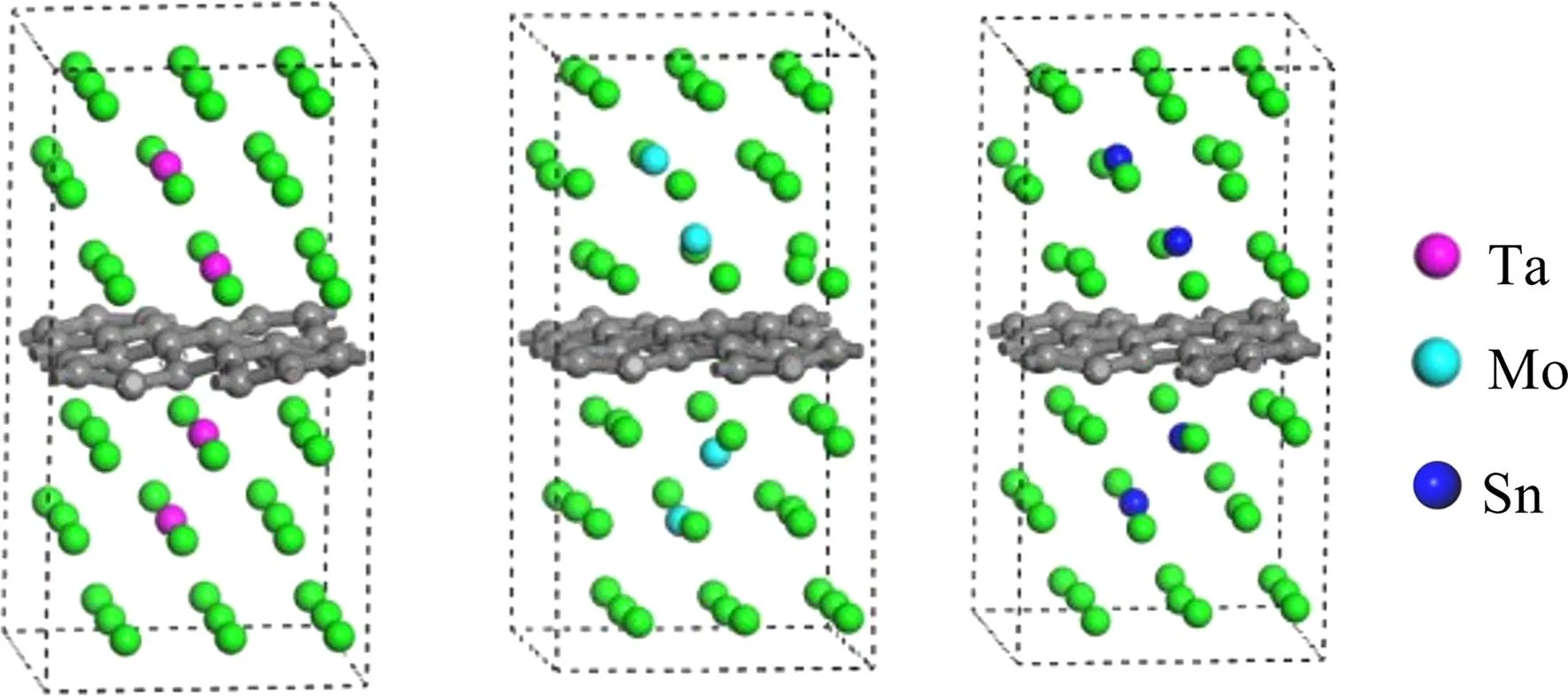
3.2 Ta元素掺杂界面电子性质

3.3 Mo元素掺杂界面电子性质

3.4 Sn元素掺杂界面电子性质


4 结 论
猜你喜欢
高中数理化(2023年6期)2023-08-26
辽宁科技大学学报(2022年5期)2023-01-04
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
中学生数理化·中考版(2021年10期)2021-11-22
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27
原子与分子物理学报(2020年5期)2020-03-17
考试周刊(2018年39期)2018-04-19
新高考·高一物理(2015年6期)2015-09-28
