
远端遗传性运动神经病的临床特征及DNAJB2基因型研究
2024-04-08 01:19:56李茂华
重庆医学 2024年6期
李茂华,王 玥
(陆军军医大学第二附属医院神经内科,重庆 400037)
远端遗传性运动神经病(distal hereditary motor neuropathies,dHMN),又称远端脊髓性肌萎缩症(distal spinal muscular atrophy,dSMA),是一种遗传异质性疾病。dHMN最常见的特征是缓慢进行性的远端肢体肌肉无力和萎缩,罕有感觉累及[1]。迄今为止,已发现30多个与dHMN相关的致病基因[1-2]。在不同的研究中,dHMN的遗传诊断率和遗传分布各不相同。LIU等[3]的研究显示我国华北地区该病诊断率为39%,来自英国的3项研究报道的诊断率分别为14.3%、26.4%和32.5%[4-6]。dHMN总体分子诊断率相对较低,表明存在更多的致病基因尚未确定,dHMN是遗传性周围神经病变最罕见的亚型之一,进一步了解其临床特征和遗传特点,有助于更好地了解该疾病。现回顾性分析1 例DNAJB2基因变异致dHMN患者的临床资料,并结合相关文献及已报道病例进行讨论。
1 临床资料
患者,男,31岁,因进行性双下肢无力、肌肉萎缩7年于2021年7月26日入院。患者7年前无明显诱因出现跑步费力,5年前逐渐出现站立下蹲后双下肢无力,2年前开始出现下肢行走不协调、拖曳,右下肢更为明显,之后发现右侧小腿逐渐变细,症状逐渐加重,出现双侧小腿肌肉萎缩,行走时易跌倒,双下肢踝关节疼痛,双侧髋关节疼痛,伴左下肢外侧皮肤麻木,双侧臀部肌肉跳动,认知语言正常,病程中无大小便失禁,无吞咽困难,无饮水呛咳,无听力下降。患者为足月顺产,自幼生长发育正常。父母体健,近亲结婚,1个妹妹1个哥哥体健,1个儿子1个女儿体健。神经系统查体:神清,对答切题,定向力、计算力正常,颅神经检查无异常,双侧腓肠肌、胫前肌及双足肌萎缩,四肢肌张力正常,双上肢肌力5级,双下肢近端肌力4级,远端肌力2级,无高弓足或扁平足。行走时双足下垂,不能垫脚及脚跟行走,下蹲不能站立,双上肢腱反射(+++),双侧膝反射(++),双侧踝反射未引出,双下肢病理征阴性。双侧深浅感觉正常。
辅助检查:血常规、肝功能、肾功能、电解质、抗核抗体谱、抗心磷脂抗体、红细胞沉降率、凝血功能均无异常。腰椎穿刺压力、脑脊液常规及生化检查未见明显异常。磷酸肌酸激酶(CK)196.2 IU/L(参考值26~174 IU/L)。心电图、心脏彩超未见明显异常。下肢血管彩超:右侧股总动脉粥样硬化斑块形成。腹部彩超:双肾肾窦反射稍增强,肝胆胰脾二维及彩色多普勒超声未见异常。颈段、胸段、腰段脊髓MRI:未见明显异常。神经传导速度:双侧腓神经、胫神经波幅减小,双侧腓神经运动传导速度减慢,左侧腓浅神经感觉神经波幅及传导速度稍降低。双下肢腓肠肌H波、F反射均未引出。肌电图:(右侧腓肠肌记录)插入电位明显延长,未见失神经电位;主动用力差,可见运动单位减少,平均时限延长。(左侧腓肠肌记录)插入电位延长,未见失神经电位;轻用力时运动单位电位平均时限稍延长、电压正常,多项波增多,重用力呈干扰相。双下肢肌肉MRI:双侧大腿、小腿肌群信号不均,压脂序列呈高信号影,存在轻微脂肪浸润和水肿(图1)。肌肉活检:左股四头肌活检,HE染色显示肌纤维大小不等,部分肌纤维萎缩,见小群萎缩、核内移和核聚集,见个别肌裂和镶边空泡(图2A、2B),ATPase染色可见Ⅱ型肌纤维优势及群组化趋势(图2C);Dysferlin染色显示肌纤维膜及胞质阳性(图2D)。
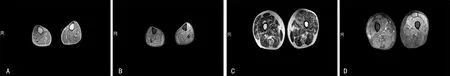
A、B:双侧小腿肌群信号不均,压脂序列高信号影,存在轻微脂肪浸润和水肿;C、D:双侧大腿肌群信号不均,压脂序列高信号影,存在轻微脂肪浸润和水肿。
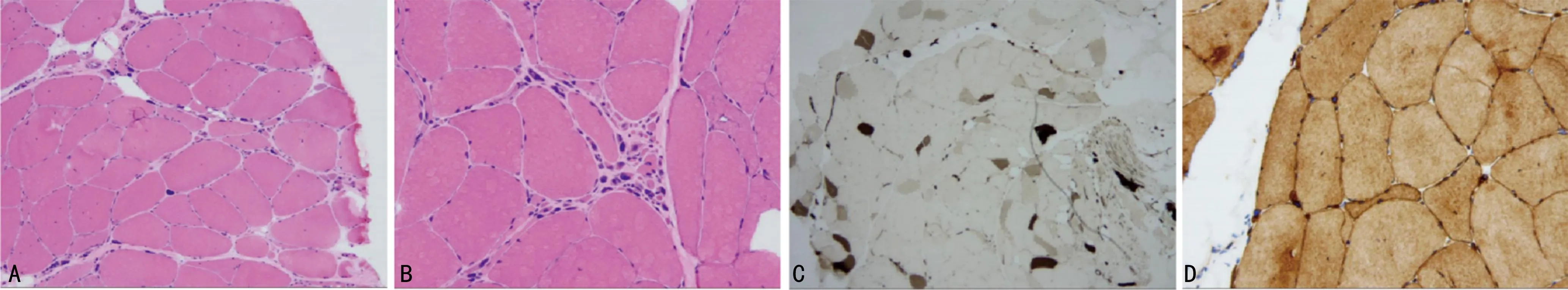
A、B:HE染色显示肌纤维大小不等,部分肌纤维萎缩,见小群萎缩、核内移和核聚集,见个别肌裂和镶边空泡;C:ATPase染色可见Ⅱ型肌纤维优势及群组化趋势;D:Dysferlin染色显示肌纤维膜及胞质阳性。
全外显子组测序及Sanger测序检测到该患者DNAJB2基因(NM-001039550.1)chr2:220145325,c.91C>T(p.H31Y)纯合变异,编码区第91位碱基由C变异成T,导致编码蛋白的第31位氨基酸由组氨酸变异成酪氨酸。经Sanger验证,受检者父母杂合携带该变异(图3)。该变异未见文献报道,该变异未在正常人群数据库中检出(PM2-Supporting),生物信息分析软件SIFT、PolyPhen2-HVAR和Mutation-Taster均预测有害(PP3)。根据现有证据,该变异定义为意义未明变异(PM2-Supporting+PP3)。予以B族维生素营养神经、康复理疗,患者病情无改善。
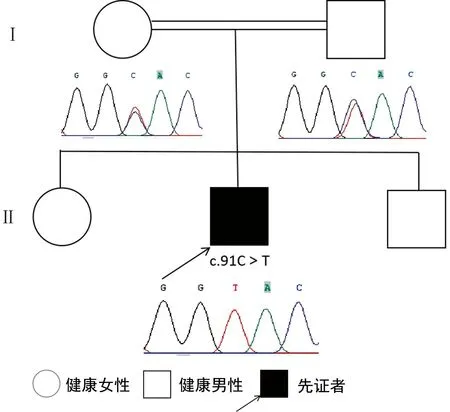
先证者DNAJB2基因存在c.91C>T点突变(p.H31Y),受检者父母杂合携带该变异,表型正常。
2 讨 论
dHMN过去被认为是一组单纯的运动神经病变,其特征是脊髓运动神经元的进行性退行性变,表现为四肢远端肌肉无力和萎缩。通常发病于儿童或青年时期,其病程进展缓慢。最初表现为下肢远端缓慢进行性无力,然后逐渐累及腿部近端肌肉,甚至影响到手部肌肉。目前发现某些类型的dHMN后期可伴有亚临床感觉异常、锥体征象和其他神经系统症状[4]。此外,dHMN与其他退行性疾病,如腓骨肌萎缩症(charcot-marie-tooth,CMT)、肌萎缩性侧索硬化症(amyotrophic lateral sclerosis,ALS)、遗传性痉挛性截瘫、脊髓小脑共济失调和肌病之间存在临床重叠[7]。到目前为止,已发现超过30个与dHMN有关的致病基因[4]。这些基因编码的蛋白参与细胞内各种生化活动,如轴突运输(HSP1、HSPB3、HSP8、DNAJB2、DCTN1、SYT2、PLEKHG5)、tRNA氨基酰化(AARS1、GARS1、HARS1、WARS1)、RNA代谢和DNA完整性(FBXO38、SETX、IGHMBP2)、离子通道和转运体(TRPV4、SLC5A7、SLC12A6、ATP7A)、内质网(REEP1、BSCL2、SIGMAR1)等[8]。大量与dHMN和相关神经肌肉疾病相关的基因具有影响轴突运输的潜在病理机制。dHMN最常见的遗传原因是HSPB1、GARS1、BICD2和DNAJB2基因的突变,而3.1%的患者携带SORD双等位基因突变。DNAJD2相关的dHMN患者在西班牙队列中相对常见[9]。大多数dHMN患者表现为常染色体显性遗传,常染色体隐性遗传dHMNs与SORD、IGHMBP2、PLEKHG5、DNAJB2、SIGMAR1、SYT2、ATM、TBCE和VRK1基因的变异相关[10-11]。
目前根据临床表型和基因表型将dHMN分为单纯的dHMN、伴有轻微感觉受累的dHMN和dHMN+(dHMN合并其他神经功能受累)[3]。dHMN和CMT之间有明显的重叠,一个基因的相同突变可以导致这两种表型。两种疾病共享许多致病基因(如HSPB1、HSPB27、BSCL2、DCTN1)[3],它们代表了从单纯运动神经病变到运动和感觉神经病变的连续体[1]。FRASQUET等[12]对来自西班牙5个不同家族的10例携带DNAJB2 c.352+1G>A突变的患者进行长期随访观察发现,DNAJB2 c.352+1G>A突变可表现为dHMN和CMT2,一些具有初始dHMN表型的病例演变为CMT2表型。此外,HSJ c.352+1G>A突变患者中1例发展为青年发病的帕金森病(parkinson disease,PD),另1例患者发展为额颞叶痴呆,在随访时间最长的患者中(症状出现后30余年),有患者最终进展为ALS[12]。在一个具有DNAJB2 c.352+1G>A突变的巴西家族中,个体还表现为PD和小脑性共济失调[13]。可能与DNAJB2阻止与ALS相关的SOD1突变蛋白的聚集[14],以及与PD相关的Parkin突变蛋白的聚集相关[15]。
DNAJB2最初被描述为热休克蛋白J1(Heat-Shock Protein J1,HSJ1),属于J蛋白的Ⅱ类(DNAJB亚家族),是一种参与泛素-蛋白酶体系统相关降解的共同伴侣蛋白,从而保护神经元免受细胞毒性蛋白聚集[16-17]。DNAJB2包含N末端J结构域、富含G/F的区域,选择性剪接产生两种不同的蛋白质异构体,并表现出不同的亚细胞定位:DNAJB2a(HSJ1a含有277个氨基酸,31×103)定位于细胞质和细胞核,而DNAJB2b(HSJ1b含有324个氨基酸,36×103),具有更长的C端,通过C端香叶基香叶基化而锚定在内质网的细胞质面,主要定位在核周[18]。DNAJB2在神经元、大脑和视网膜的神经元层、运动神经元和神经轴突中高表达,与轴突神经丝共定位,DNAJB2突变可能损害轴突运输,并存在于成熟肌肉中神经肌肉接头的突触后部分[19]。DNAJB2双等位基因缺失导致的主要临床表型是外周轴突神经病变。DNAJB2的突变常为隐性遗传,导致异常剪接。一个具有dHMN表型的家族显示纯合子剪接位点突变c.229+1G>A[20],导致DNAJB2信使RNA中内含子4保留,存在过早终止密码子,蛋白表达缺失。DNAJB2 c.352+1G>A突变已被报道为dHMN的致病突变[14],c.352+1G>B导致内含子保留、翻译过早终止和DNAJB2蛋白表达减少,导致DNAJB2功能的丧失。DNAJB2纯合子剪接c.163-3C>G突变可表现为dHMN和锥体束体征[3]。
本例患者24岁起病,以下肢远端进行性肌无力、萎缩为主,与之前报道的DNAJB2突变的dHMN表型一致。神经传导速度检测提示双侧腓神经、胫神经波幅减小,双侧腓神经运动传导速度减慢,双下肢腓肠肌H波、F反射均未引出。肌电图提示神经源性改变,肌肉活检伴神经源性肌损害。但患者双上肢腱反射活跃,左侧腓浅神经感觉神经波幅减小且传导速度稍降低。基因检测发现DNAJB2 c.91C>T新发纯合变异,在家系分析中与疾病表型共分离,临床意义尚不明确,后续需功能验证评估突变的致病性。根据临床病史、检查及基因检测,诊断为DNAJB2基因突变相关的dHMN。患者肌肉磁共振提示轻微脂肪浸润和水肿,CK轻度升高,CK升高可能与神经源性损害的去神经支配导致肌膜完整性损伤相关[21]。患者双下肢远端肌无力伴萎缩合并上运动神经元和轻微的感觉神经受累症状,这在DNAJB2突变的患者中很少被描述,这可能会进一步扩大DNAJB2突变的表型和基因谱。
dHMN不仅和CMT2在临床和遗传学方面有较多的重叠,dHMN患者还携带与遗传性痉挛性截瘫、ALS和脊髓性肌萎缩症(FUS、KIF5A、KIF1B、ZFYVE26、DNAJB2)相关的基因变异[3]。目前,只有15.0%~32.5%的dHMN患者具有基因特征[9],有必要进行系统的研究,以更好地描述dHMN的人群分布,采取多种基因检测手段联合分析有助于提高遗传诊断。
猜你喜欢
中国现代医生(2022年21期)2022-08-22 03:30:42
中国临床医学影像杂志(2022年6期)2022-07-26 07:17:24
中国临床医学影像杂志(2022年5期)2022-07-26 07:11:54
国际放射医学核医学杂志(2021年10期)2021-02-28 08:43:54
现代园艺(2017年21期)2018-01-03 06:41:32
中国医药科学(2017年9期)2017-08-04 21:42:14
放射学实践(2016年6期)2016-12-15 21:55:30
天津农学院学报(2016年2期)2016-12-01 05:40:05
西南医科大学学报(2016年4期)2016-01-03 01:26:36
中国康复理论与实践(2015年10期)2015-12-24 05:42:44
