
电感耦合等离子体质谱法测定大批量钼多金属矿中钼及4种主要伴生元素的含量
2024-04-02 07:22李俊东黄青春何文江左修源
理化检验-化学分册 2024年3期
贾 雷,李俊东,黄青春,祁 伟,何文江,左修源
(1.中国地质调查局呼和浩特自然资源综合调查中心,呼和浩特 010010;2.营口理工学院 化学与环境工程学院 辽宁省化学助剂合成与分离重点实验室,营口 115014)
Mo具有耐高温强度、耐磨性和抗腐蚀性等优点,在我国的储量位于世界前列,且开发条件便捷、品位高,被广泛用作生产各种合金钢或高级合金的添加剂。Mo多数以硫化物的形态存在,其主要分布在辉钼矿中,常与Cu、W、Zn、Ag等元素伴生[1]。
钼多金属矿的分析不仅对地质勘探中矿区内具层状特征、矿化特点后期演变、成矿规模和成矿机制具有重要意义,还对矿石的冶炼、提取、分离等工艺过程起着至关重要的作用[2-3]。目前,测定钼多金属矿中Mo、W、Cu、Zn和Ag的分析方法主要为重量法[4]、容量法[5]、原子吸收光谱法[6-7]和极谱法[8],这些方法前处理繁琐、需添加试剂较多、成本高,难以适用大批量快速测定。电感耦合等离子体原子发射光谱法(ICP-AES)作为常量元素分析的重要手段,因其线性范围宽、可测定高浓度水平而被广泛使用。电感耦合等离子体质谱法(ICP-MS)对常量元素的分析报道少之又少,其主要存在如下问题:在高浓度水平下,质谱检测器的信号会不会饱和漂移;长期接收强信号,检测器的寿命会不会受影响;如果检测器可以在高浓度水平下运行,其稳定性能否保证等[9-12]。
为了降低或消除上述影响,本工作选择在250 ℃电热板上用聚四氟乙烯坩埚敞口酸消解样品18 h,利用全自动样品稀释系统稀释样品母液100倍,增大ICP-MS稀释气流量以降低物理干扰,通过10 h仪器稳定性试验的研究,提出了ICP-MS快速、准确测定大批量钼多金属矿中Mo、W、Cu、Zn和Ag含量的方法,满足生产和科研要求。
1 试验部分
1.1 仪器与试剂
NexION 350XL型电感耦合等离子体质谱仪;LabTech EG35A plus型温控电热板;Saxillex DST-1000型酸纯化器;Vulcan42S型全自动样品前处理工作站;Milli-Q Advantage A 10型超纯水设备;FA1204B型电子天平。
调谐溶液:以5%(体积分数)硝酸溶液为基底,Be、Ce、Fe、In、Li等元素的质量浓度均为1 mg·L-1的混合溶液。
Rh标准储备溶液:1 000 mg·L-1。
Rh内标溶液:10 mg·L-1,移取1 mL 1 000 mg·L-1的Rh标准储备溶液至100 mL容量瓶中,用水稀释并定容,混匀,配制成质量浓度为10 mg·L-1的Rh内标溶液。
Cu、Zn、Ag元素的混合标准储备溶液:10 mg·L-1。
Mo、W元素的混合标准储备溶液:10 mg·L-1。
混合标准溶液系列:取适量的Cu、Zn、Ag元素的混合标准储备溶液和Mo、W元素的混合标准储备溶液,用水逐级稀释,配制成质量浓度为0,0.10,0.20,0.50,1.00 mg·L-1的混合标准溶液系列。
氢氟酸、硝酸、高氯酸、盐酸、硫酸、磷酸为优级纯;试验用水为超纯水(电阻率18.2 MΩ·cm);钼矿石国家标准物质GBW 07142、GBW 07143、GBW 07144,来自地球物理地球化学勘查研究所。
1.2 仪器工作条件
PFA-ST3型微流量雾化器,石英材质矩管和雾化室;等离子体气流量 16.0 L·min-1,雾化气流量 0.96 L·min-1,辅助气流量 0.95 L·min-1,稀释气流量 0.6 mL·min-1;射频(RF)功率 1 600 W;测量模式为动能歧视(KED)模式,扫描方式为跳峰,扫描10次,重复测定3次,测定时间 2 min,分析时间 2.8 min。
1.3 试验方法
准确称取0.05 g(精确至0.000 1 g)样品于聚四氟乙烯坩埚中,将坩埚均匀隔开放在电热板上,用少量水润洗,加入4 mL硫酸和8 mL硝酸的混合溶液,静置5 min,使样品与溶液混合均匀,将电热板温度调至250 ℃,加热18 h蒸干溶液。待冷却至室温,加入3~4 mL的10%(体积分数)硝酸溶液将其溶解,转移至25 mL容量瓶中用水定容,作为样品母液。用Vulcan42S型全自动样品前处理工作站稀释样品母液100倍后直接按照仪器工作条件测定。
2 结果与讨论
2.1 消解方法的选择
常用的消解方法主要有微波消解、密闭高温高压消解、敞口溶样消解。其中,微波消解具有取样量大、溶剂量少、酸消解效率高和反应时间短等优点,但是存在对酸反应剧烈的样品容易发生爆炸、样品处理数量有限、成本高、对于某些样品不易消解、在高温高压下开盖过滤导致刺激性酸雾释放引起人体不适等缺点。密闭高温高压消解具有可降低元素损耗、取样量小、成本低、安全性高等优点,但存在耐高温性能差、密封性差容易导致泄漏等缺点[13-14]。对于大批量科研和生产活动,试验选择在电热板上用聚四氟乙烯坩埚敞口酸消解的方式,该方法具有操作简单、多酸混合能有效消解样品、样品处理量大等优点。
2.2 溶样体系的选择
试验采用溶样体系1~7(氢氟酸6 mL+硝酸3 mL+高氯酸1 mL、氢氟酸5 mL+盐酸5 mL、氢氟酸8 mL+硝酸4 mL、硫酸8 mL+磷酸4 mL、硫酸4 mL+硝酸8 mL、氢氟酸5 mL+硫酸3 mL+硝酸2 mL、盐酸9 mL+硝酸3 mL)对国家标准物质GBW 07144消解,其中Mo、W、Cu、Zn、Ag的认定值分别为5.08×106,732,266,68,2.1 mg·kg-1,消解结果见表1。其中,Mo测定时稀释1 000倍,其余元素测定时稀释100倍,测定值为稀释后结果;溶样体系4的消解温度为190 ℃,其余均为250 ℃。
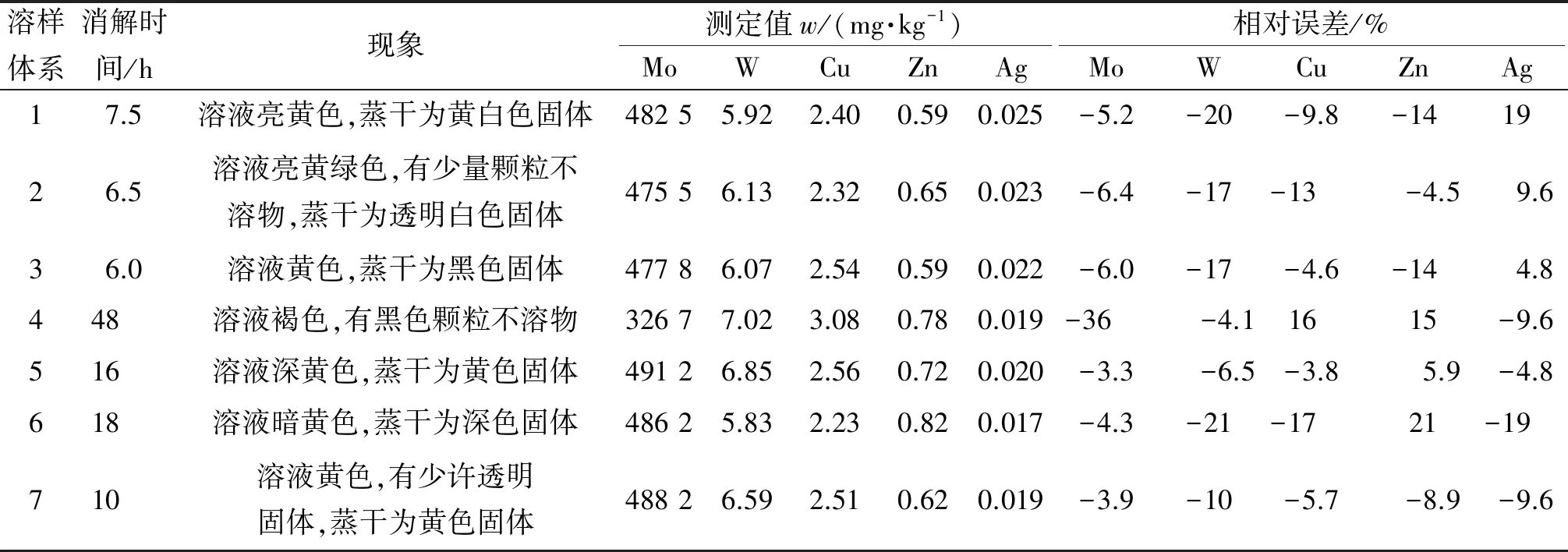
表1 溶样体系对消解结果的影响
结果表明:银离子与氯离子存在沉淀溶解平衡产生氯化银沉淀,可溶性银离子在氯离子的溶液里难以长时间稳定存在,因此溶样体系中含有盐酸时难以得到Ag的准确结果;硫酸-磷酸体系对W具有分解快速的特点,但在190 ℃电热板上敞口消解48 h,溶液蒸发缓慢,有难溶黑色颗粒物,磷酸黏度高容易对管路产生堵塞,并且难以适应大批量生产要求;氢氟酸-硝酸体系对于大多数元素溶出效果较好,但会引入F、N这类同位素干扰;高氯酸使用过多会在三级锥口累积成盐,有爆炸风险。从消解结果可知,硫酸-磷酸体系所得W测定结果较为准确,相对误差绝对值较小;氢氟酸-盐酸体系对Zn溶出效果较好,测定结果准确;测定元素相对误差绝对值最大的是氢氟酸-硫酸-硝酸三酸体系,最小的是硫酸-硝酸体系。综合消解时间、酸用量和消解结果,试验选择硫酸4 mL+硝酸8 mL作为溶样体系。
2.3 稀释方式的选择
将未知样品先通过X-荧光分析仪大致确定待测元素的含量范围,钼多金属矿中Mo、W、Cu、Zn的质量分数在1.0×105mg·kg-1内,Ag的在10~600 mg·kg-1内,此含量范围远超过ICP-MS的检测范围[最高质量浓度为10 mg·L-1(5 000 mg·kg-1)],因此确定稀释100倍再进行测定,将高浓度水平样品稀释可降低质谱检测器的强信号,削弱多原子光谱干扰强度。手动稀释大批量样品可造成稀释倍数的误差,增加试剂使用量,配制时间长、容易引起污染;逐级稀释法能够获得一系列不同稀释比例的溶液,通过观察基体信号强度和质量浓度的线性关系,得到最小基体影响的稀释倍数,但对于大批量的生产操作繁琐、费时。试验选择操作简单、易行、经济的Vulcan42S型全自动样品消解稀释系统,设置稀释倍数、加液速率、定容高度等仪器参数进行自动稀释。
2.4 消解温度和消解时间的选择
试验以钼矿石国家标准物质GBW 07142为研究对象,其中Mo、W、Cu、Zn、Ag的认定值分别为1 500,518,46,365,0.1 mg·kg-1。以硫酸-硝酸体系消解,以30,60 ℃为间隔段改变消解温度,采用电热板敞口消解方式,经相关系数修约,结果见表2。其中:测定Ag时不稀释样品母液;测定值为稀释后结果。
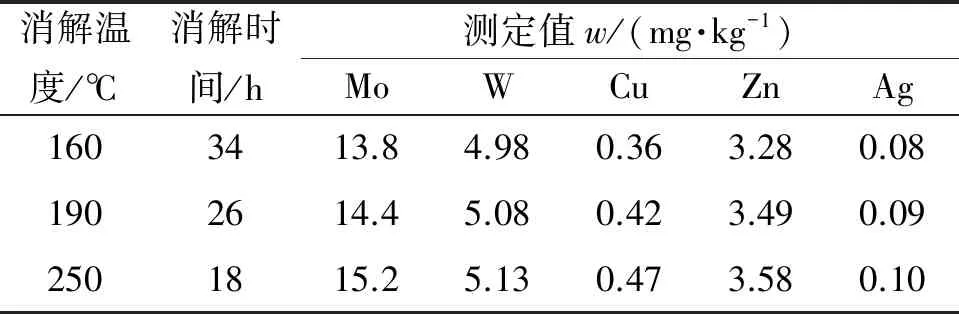
表2 消解温度和消解时间对测定结果的影响
结果表明,样品在160 ℃、34 h即消解完成,由于各元素对反应温度不同的适应性,为了样品被彻底分解,各元素损失降低至最小,试验选择250 ℃为消解温度,18 h为消解时间。
2.5 物理干扰
对于高浓度水平样品,物理干扰不可避免,其易引起检测信号增强、精密度差、回收率低等问题,对ICP-MS仪器寿命有损伤,并与稀释气流量和雾化效率有关。利用稀释气进行高浓度水平样品的稀释,使整个气溶胶的含水量和酸量降低,削弱电离能过大元素的检测信号,进而对仪器起到保护作用。因此,试验采用0.6 mL·min-1的稀释气流量以降低物理干扰。
2.6 质量数和丰度的选择
ICP-MS的质谱干扰主要有4种:(1)同量异位离子干扰,即质量几乎相同的两种不同元素的同位素所造成的干扰;(2)仪器和试样制备所引入的杂质离子干扰,即采样锥和分离锥材料中溅出的金属离子以及试剂和水中微量杂质离子所造成的干扰;(3)多原子分子离子干扰;(4)氧化物和氢氧化物离子干扰。对待测元素影响较大的是同位素干扰,选择同位素的基本原则是在灵敏度允许条件下尽量避开干扰,选丰度大的同位素。试验选择Mo、W、Cu、Zn、Ag的质量数和丰度分别为97.905 5和24.13%、183.951 0和30.64%、62.929 8和69.17%、106.905 0和51.84%、65.926 0和27.90%。
2.7 内标的选择
Rh作为内标可以同时校正多种元素,校正基体干扰作用强,不受待测元素质量数差异和电极电位差异的影响,内标校正主要用于检测和校正信号的短期漂移和长期漂移,并可校正一般的样品基体影响。内标选择的原则如下:该元素必须有一定的浓度水平,其产生的信号强度不应该受到计数统计的限制,待测样品中没有这个元素为最佳,它的质量和电离能应与待测元素接近,尽可能确保所配制的内标元素浓度水平下所产生的信号和待测元素一致。综合考虑以上因素,试验选择Rh作为内标。
2.8 标准曲线、检出限和测定下限
按照仪器工作条件测定混合标准溶液系列,以各元素的质量浓度为横坐标,对应的响应值为纵坐标绘制标准曲线。结果表明,各元素的质量浓度在1.00 mg·L-1内与对应的响应值呈线性关系,所得线性回归方程和相关系数见表3。
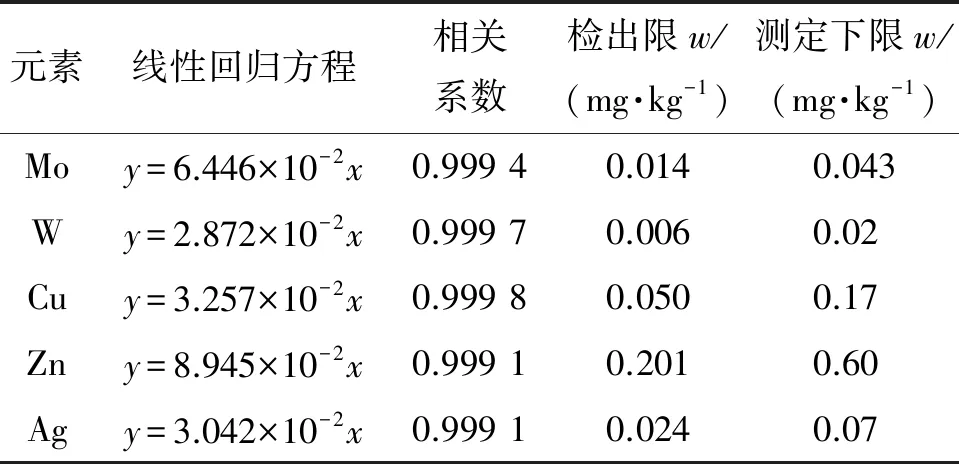
表3 线性参数、检出限和测定下限
在优化的试验条件下,连续测定12份空白样品溶液,以响应值的3倍标准偏差s计算仪器的检出限(3s),以响应值的10倍标准偏差计算仪器的测定下限(10s),结果见表3。
结果表明:各元素的检出限为0.006~0.201 mg·kg-1,样品和标准物质需稀释到相应倍数以适应检出限范围和满足测试要求。
2.9 精密度和准确度试验
按照试验方法分析钼矿石国家标准物质GBW 07142、GBW 07143,各称取4份样品平行测定8次,计算测定值的相对标准偏差(RSD),结果见表4,测定值和认定值均为稀释后结果。
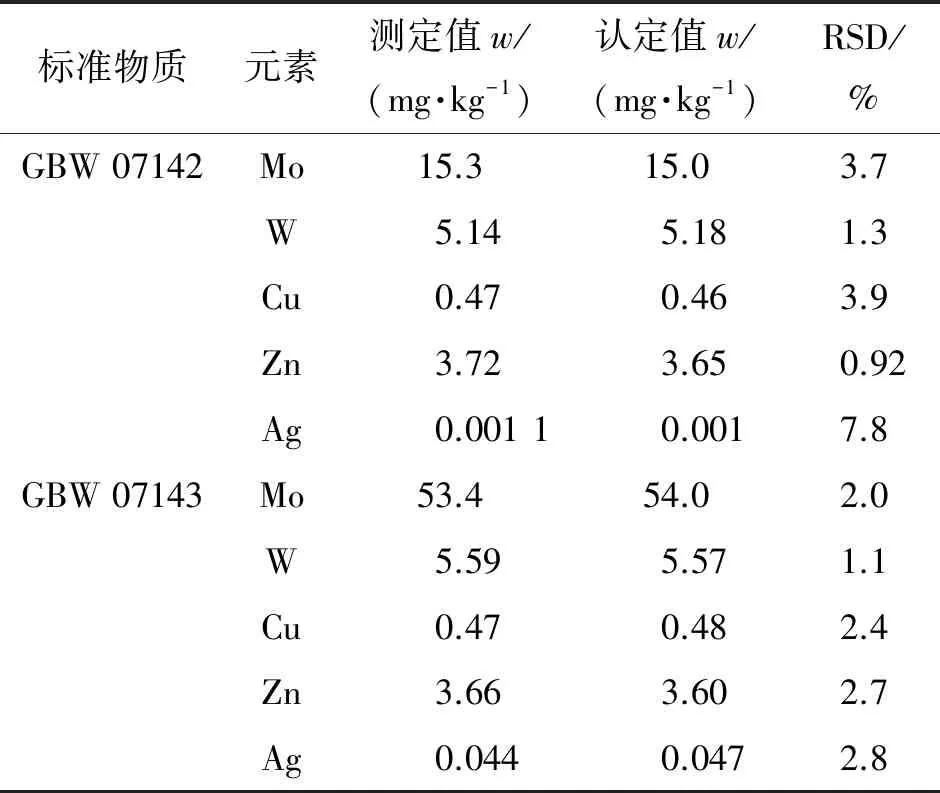
表4 精密度和准确度试验结果(n=8)
结果表明:两种钼矿石国家标准物质的测定值和认定值基本一致,测定值的RSD均小于8.0%,说明方法准确度和精密度良好。
2.10 仪器稳定性试验
高浓度水平样品在测试时间较长情况下会引起仪器信号漂移,导致精密度下降。试验选定10 h为工作时长,探究在以Rh为内标的条件下仪器信号的漂移情况,以评价100个样品测试结果是否具有稳定性,在国家一级标准物质GBW 07142中加入1.00 mg·L-1混合标准溶液,连续测定10 h,考察仪器的稳定性。结果表明:监测前3 h时,5种元素的加标回收率稳定,仪器性能良好;在监测第8 h时,5种元素的加标回收率出现明显降低,仪器的信号漂移,测定值的RSD偏大;重新更换蠕动泵管卡,清洗锥孔,重新校正标准曲线后,每种元素测定值的RSD均小于15%,加标回收率为87.9%~93.7%。因此,在进行大批量长时间测试时,需及时重新更换零件和校正标准曲线,才能满足测试要求。
2.11 回收试验
对钼矿石国家标准物质GBW 07142、GBW 07143进行加标回收试验,加标量约为2.9节测定值,计算回收率,结果见表5。
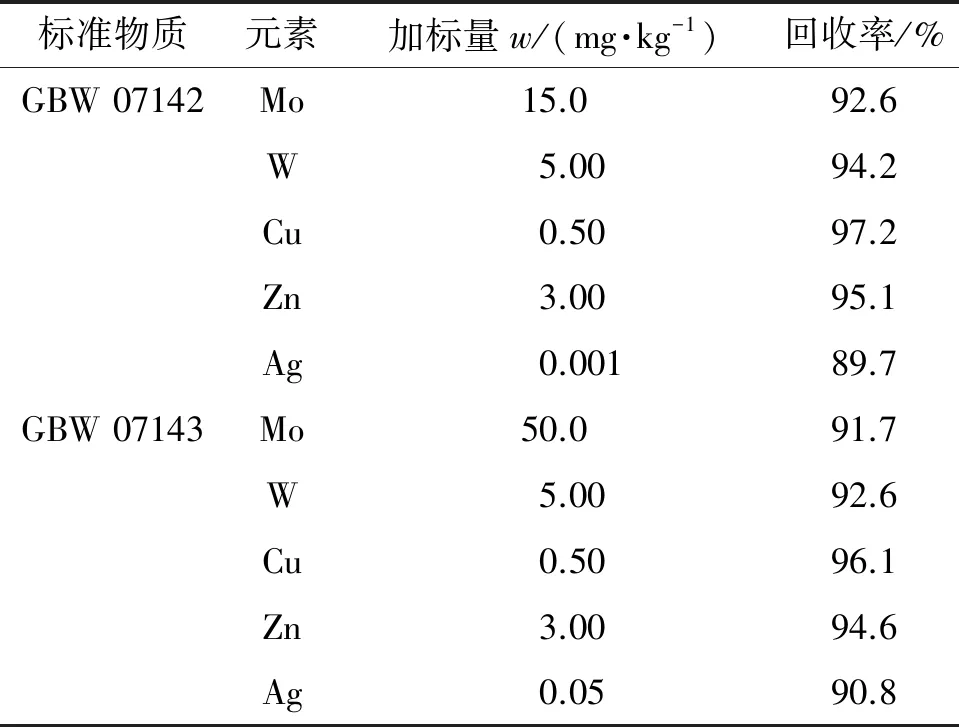
表5 回收试验结果
2.12 样品分析
按照试验方法分析100个样品,结果显示,Mo的检出量为1.90×102~7.93×103mg·kg-1,W的检出量为7.10×102~1.95×104mg·kg-1,Cu的检出量为6.16×102~3.00×104mg·kg-1,Zn的检出量为6.30×102~2.15×104mg·kg-1,Ag的检出量为1.00×10~5.10×102mg·kg-1,已经达到成矿要求。
采用ICP-MS可对常量Mo、W、Cu、Zn、Ag同时分析,但微量元素分析仪器并不能长久测定高含量元素,其对检测器的影响是巨大的,需更新设备或引入辅助设备进行测试。本工作对比了用7种溶样体系消解不同时间下的结果相对误差,得到最佳溶样体系;考察了不同元素在不同温度下的消解效果,选择250 ℃、18 h为溶样条件;充分考虑大规模生产、科研的成本和测定的准确度,选择敞口酸溶的消解方式;利用全自动稀释系统降低了高浓度水平样品的手动稀释误差;增大稀释气流量以降低高浓度水平样品的物理干扰;长时间测试样品时需重新校正标准曲线,以优化仪器的稳定性,得到准确结果。本方法节约了人力成本,适合对大批量、有成本控制需求的样品进行快速准确地测定。
猜你喜欢
云南化工(2021年7期)2021-12-21
中学生数理化·高一版(2020年11期)2020-12-14
科学技术创新(2020年33期)2020-11-27
冶金与材料(2019年4期)2019-09-07
中国住宅设施(2017年6期)2018-03-07
电镀与环保(2017年2期)2017-05-17
湖南大学学报·自然科学版(2016年5期)2016-06-07
中国资源综合利用(2016年6期)2016-01-22
石油化工应用(2015年10期)2015-12-24
地球化学(2015年3期)2015-07-02
