
少见子宫间叶源性肿瘤病理诊断的新进展
2024-04-01 07:59李慧,毕蕊
临床与实验病理学杂志 2024年3期
李 慧,毕 蕊
随着分子病理检测技术的不断发展,研究人员对各种子宫间叶源性肿瘤的认识进一步深化。WHO(2020)女性生殖系统肿瘤(简称第五版WHO)对子宫间叶源性肿瘤的分类、分级进行更新和补充,本文根据第五版WHO分类并结合最新研究进展,对原发于子宫和近年新发现的几种少见间叶源性肿瘤的特点进行归纳和总结,旨在为临床、病理医师的诊断和鉴别诊断提供新思路。
1 平滑肌源性肿瘤
平滑肌源性肿瘤的亚型繁多,本文重点介绍新近引起重视的几种特殊类型的肿瘤。
1.1 延胡索酸水合酶(fumarate hydratase, FH)缺陷型子宫平滑肌瘤FH缺陷型子宫平滑肌瘤临床罕见,约占子宫平滑肌瘤的1%。第五版WHO首次将其纳入子宫平滑肌瘤亚型,并强调FH基因的胚系或体系变异均可导致FH缺陷型子宫平滑肌瘤。FH胚系变异会导致遗传性平滑肌瘤病和肾细胞癌(hereditary leiomyomatosis and renal cell cancer, HLRCC),主要表现为多发性皮肤平滑肌瘤、多发性子宫平滑肌瘤和肾细胞癌[1]。70%~80%的患者约在30岁发生皮肤平滑肌瘤和子宫平滑肌瘤,15%~20%的患者在40岁以后出现恶性度高、侵袭性强的Ⅱ型乳头状肾细胞癌。因此,皮肤-和子宫的平滑肌瘤被认为是HLRCC患者的前哨肿瘤。
FH缺陷型子宫平滑肌瘤往往多发,镜下主要特征包括:间质内鹿角样血管(图1A);间质水肿,形成肺泡样水肿结构;散在分布奇异核细胞和多核细胞(图1B);卵圆形细胞核有时出现链状排列;可见胞质内嗜伊红包涵体;嗜酸性大核仁和核周空晕[2],瘤体周围肌层可见平滑肌瘤病样的模糊结节生长。免疫组化检测显示FH染色缺失(图1C)、2SC弥漫性颗粒状着色(图1D),分子检测示1q43杂合性缺失。FH表达缺失和2SC表达,有助于识别FH缺陷型平滑肌瘤。上述形态不仅出现于平滑肌瘤中,也可在恶性潜能未定的平滑肌肿瘤和平滑肌肉瘤中见到相似形态,并有FH缺失表达。因此,应对形态学具有提示特征的子宫平滑肌肿瘤进行免疫组化筛查,如发现FH缺失表达和(或)2SC的异常表达,建议进一步行分子检查筛选HLRCC的患者。
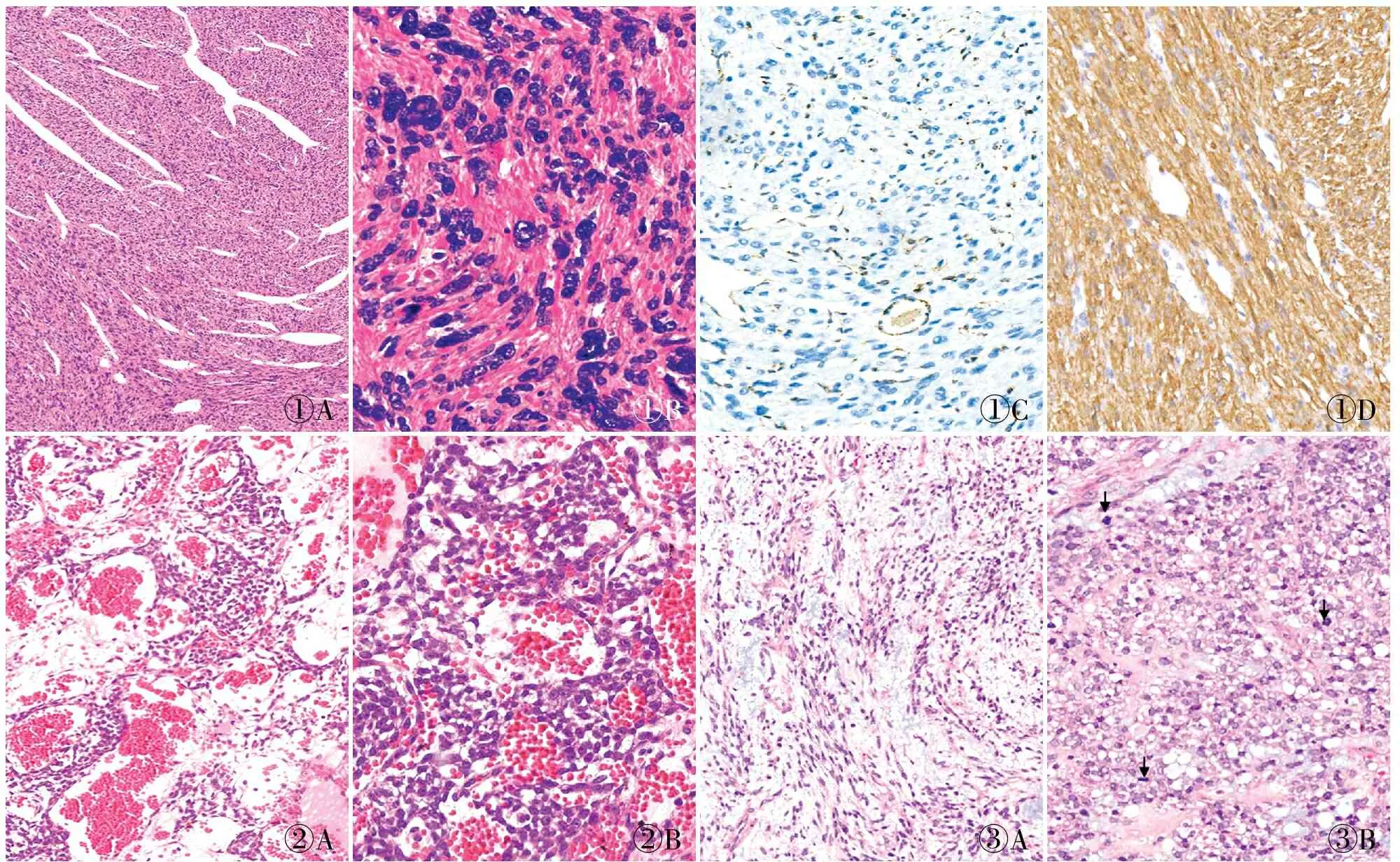
①A①B①C①D②A②B③A③B
1.2 伴PGR基因重排的上皮样平滑肌肉瘤Chiang等[3]发现在上皮样平滑肌肉瘤伴横纹肌样和梭形细胞特征时,可伴PGR基因重排,伴侣基因主要为NR4A3。患者中位年龄45岁,肿瘤呈棕色、红色或黄白色,质脆伴出血,偶可见囊性变和坏死。肿瘤位于子宫体肌层或子宫颈间质,未累及内膜。镜下呈双相性分化,上皮样或横纹肌样细胞与梭形细胞混合存在;均匀一致的上皮样细胞含丰富的嗜酸性胞质,有时胞质内可见致密的嗜酸性包涵体类似横纹肌样细胞,细胞核不规则,位于中央或偏位,偶见核沟,染色质空泡状,核仁明显,间质可见黏液样基质和微囊形态的水肿变性(图2);梭形细胞成分通常由小而温和的细胞组成,核仁不明显,细胞质丰富嗜酸呈波浪状,有时梭形细胞可呈席纹状,细胞核略拉长,核仁明显,细胞质丰富嗜酸。上皮样或横纹肌样细胞区域核分裂象活跃(约10个/10 HPF),梭形细胞区域核分裂象少见(<1个/10 HPF)。可见小动脉或大血管和脉管侵犯,坏死少见。免疫表型:desmin、ER、PR均阳性,h-Caldesmon极少阳性,CD10、HMB-45和Myogenin均阴性。伴PGR基因重排的上皮样平滑肌肉瘤患者术后均未见复发,预后较好,提示可能呈惰性生物学行为。由于文献报道的病例数有限,需积累更多数据进一步分析。
1.3 伴PLAG1基因重排的黏液样平滑肌肉瘤近年有研究发现,约25%的子宫黏液样平滑肌肉瘤伴PLAG1基因重排,伴侣基因主要为TRPS1和RAD51B[4]。患者中位年龄50岁,临床表现与普通型平滑肌肉瘤无差异,肿瘤呈结节状或弥漫性肌壁间浸润性生长,切面呈胶状,质软[5]。镜下可见肿瘤浸润性生长,梭形细胞散在分布于黏液样背景中(图3A),细胞核异型性变化较大,可轻、中或重度异型性,或偶见上皮样形态伴明显多形性;核分裂象多少不等(1~14个/10 HPF)(图3B);常见纤细的血管网和坏死;无脉管侵犯。免疫表型:肿瘤细胞弥漫表达PLAG1、SMA、desmin、h-Caldesmon,局灶表达BCOR,不表达ALK。分子检测发现黏液样平滑肌肉瘤肿瘤突变负荷较低,但拷贝数变异相对较高。文献报道黏液样平滑肌肉瘤比普通平滑肌肉瘤更具侵袭性,患者5年生存率仅11%[6]。
2 子宫内膜间质肉瘤
低级别子宫内膜间质肉瘤(low grade endometrial stromal sarcoma, LGESS)常见JAZF1∷SUZ12融合,高级别子宫内膜间质肉瘤(high grade endometrial stromal sarcoma, HGESS)有2种不同融合基因(YWHAE和BCOR),本文简要介绍BCOR基因相关的HGESS。
2.1 伴BCOR基因融合的HGESSBCOR基因异常形式主要包括BCOR基因融合和BCOR第15号外显子内部串联重复(internal tandem duplications, ITD),与伴YWHAE基因融合的HGESS相比,其侵袭性强、预后差。第五版WHO纳入了伴ZC3H7B∷BCOR 融合的HGESS,该肿瘤好发于老年女性,中位年龄54岁,肿瘤通常较大,常伴出血、坏死,患者预后较差,常伴复发、转移或死亡[7]。镜下可见梭形细胞呈杂乱的束状排列,间质内可见显著的黏液样基质(图 4A),偶可见灶区间质胶原化;细胞核呈短梭形或卵圆形,具有轻-中度异型性,核仁不明显,染色质均匀,细胞质少-中等量、嗜酸性,核分裂象易见(通常>10个/10 HPF)(图4B);可见坏死和脉管侵犯[8]。免疫表型:肿瘤弥漫表达CD10、Cyclin D1(图4C)和BCOR(图4D),不同程度表达ER和PR,局灶表达SMA和h-Caldesmon,不表达desmin。分子检测陆续发现了新的BCOR融合基因伴侣,包括EP300、NUTM2G、RGAG1、ING3、KMT2D 和CREBBP 等[9]。这些新融合的病例,与ZC3H7B∷BCOR融合病例具有相似的临床病理学特征。伴ZC3H7B∷BCOR的 HGESS常伴CDK4或MDM2基因扩增,提示CDK4抑制剂可能作为该肿瘤的潜在治疗方法[9-10]。
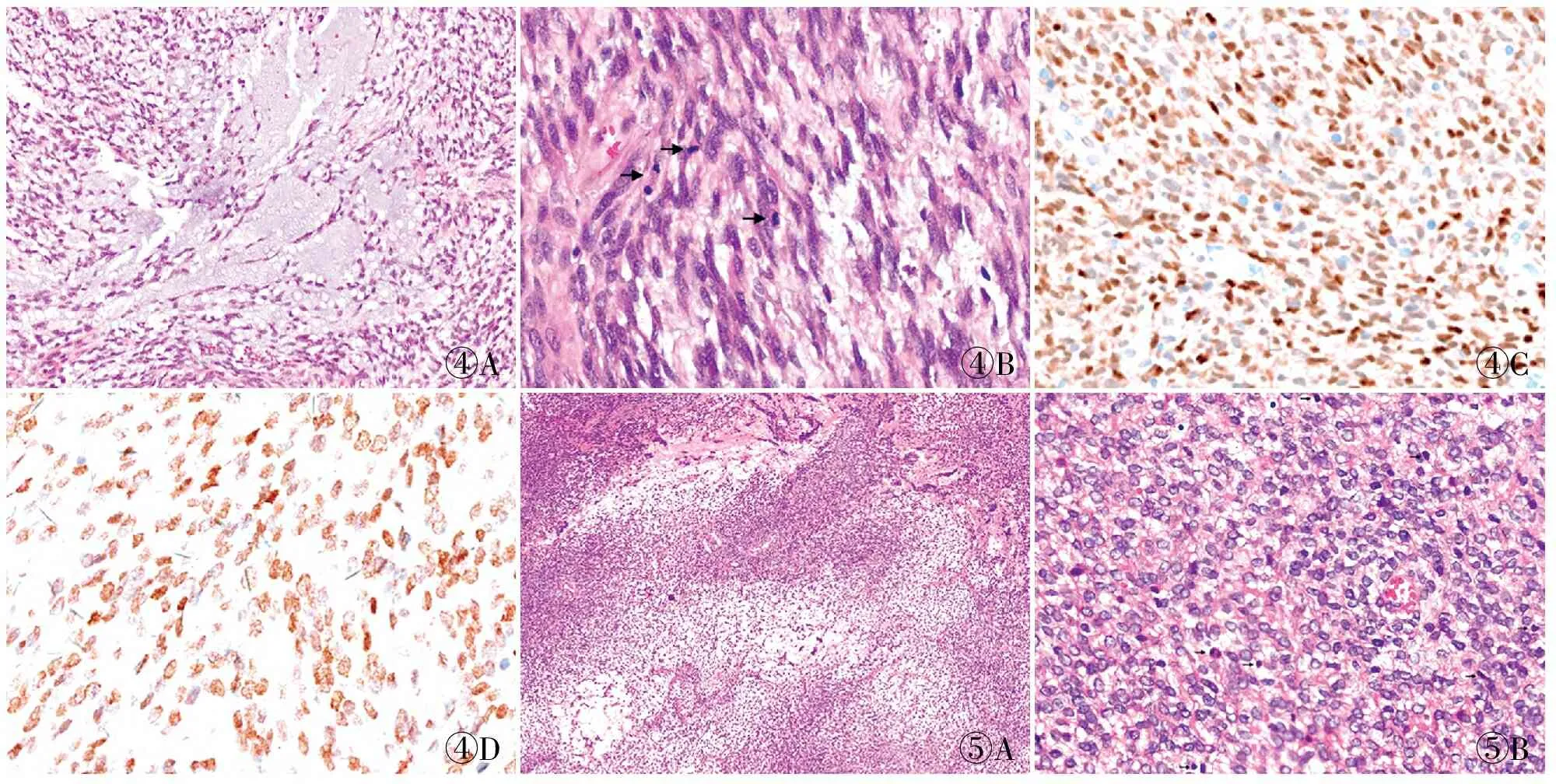
④A④B④C④D⑤A⑤B
2.2 伴BCOR-ITD的 HGESS临床显示伴BCOR-ITD的 HGESS患者相对年轻,中位年龄44岁[11-12],形态学主要包括3种成分:类似于ZC3H7B∷BCOR HGESS 的高级别梭形细胞,类似于YWHAE∷NUTM2A/B HGESS的高级别圆细胞和低级别梭形细胞[11]。中等大小的圆细胞呈巢状,偶见假乳头状或假腺样结构;核圆形或卵圆形,染色质颗粒状,核仁小,细胞质少而嗜酸性,核分裂象易见(通常>5个/10 HPF)。低级别成分类似于经典型LGESS,核分裂象少见(通常<1个/10 HPF)。间质可见黏液样或胶原变性,坏死和脉管浸润常见。免疫表型:高级别成分弥漫表达Cyclin D1和BCOR,局灶表达CD10,不同程度表达ER、PR和desmin,不表达SMA和h-Caldesmon;低级别成分也弥漫表达Cyclin D1和BCOR[10,12]。BCOR-ITD HGESS与ZC3H7B∷BCOR HGESS不同,其常伴CDKN2A/B纯合性缺失,不伴CDK4或MDM2扩增[9]。此类肿瘤需与HGESS其他亚型和子宫未分化肉瘤鉴别,上述形态特点、免疫表型和分子改变有助于诊断。
2.3 伴BCORL1基因异常的HGESSBCORL1是与BCOR同源的转录辅助抑制因子,两者在基因转录调控过程中具有相似的生物学功能,BCORL1可与BCOR互换形成PRC1复合物变异体,发挥转录抑制功能。新近报道发现HGESS也可伴BCORL1基因异常,包括JAZF1∷BCORL1、EP300∷BCORL1、BCORL1自身重排、突变和纯合性缺失[13-14]。患者中位年龄56岁,组织学、分子异常和预后均与伴BCOR基因重排的HGESS相似(表1,图5)。免疫表型:肿瘤细胞弥漫表达CD10、Cyclin D1、BCOR、ER和PR。约50%伴BCORL1基因异常的HGESS患者,也同时伴CDK4基因扩增或CDKN2A缺失,亦有少数病例伴NF1基因改变和mTOR-NF2-AKT通路异常。除CDK4抑制剂外,MEK或mTOR抑制剂也有望成为其治疗的新方法。
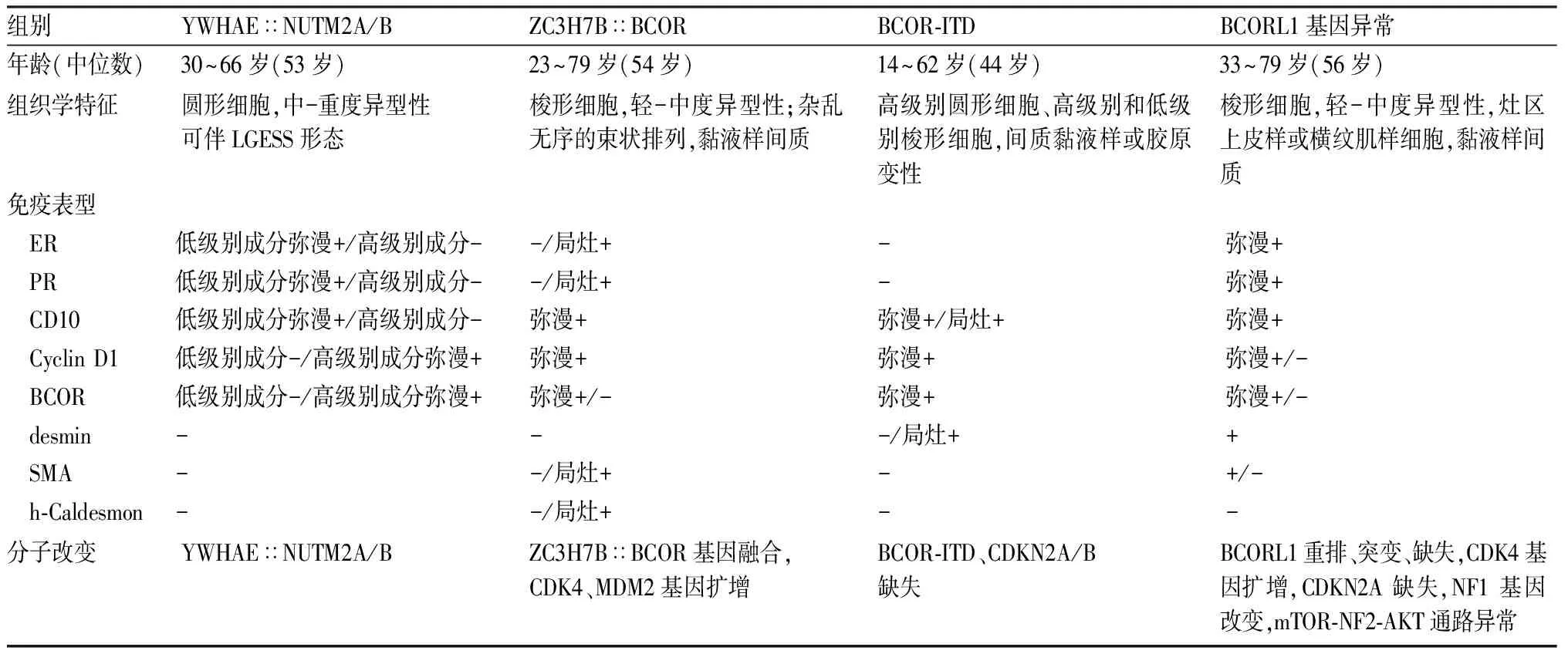
表1 不同分子亚型的HGESS临床病理特征比较
3 类似卵巢性索肿瘤的子宫肿瘤(uterine tumor resembling ovarian sex cord tumor, UTROSCT)
近年研究发现UTROSCT是一组异质性肿瘤,主要涉及NCOA1-3、GREB1和ESR1基因的融合。患者年龄范围较广(12~86岁),无特异性临床症状,可出现异常阴道出血、盆腔不适或不孕。肿瘤可位于内膜下(息肉状)、肌壁间或浆膜下,平均最大径5.1~6.1 cm,边界清楚,黄褐色或灰白色实性、质软结节,但有时边界不清,呈囊实性,出血和坏死不常见[15-17]。肿瘤含有多少不等的性索结构,包括梁状、条索、巢状、Sertoli管状和网状结构等。免疫组化提示多向分化,肿瘤细胞不同程度表达性索标志物 (Calretinin、WT-1、 inhibin和Melanin-A),上皮标志物 (CK、EMA) 和肌源性标志物(SMA、desmin和h-Caldesmon)。第五版WHO中将类似UTROSCT列为“杂类间叶源性肿瘤”,不再归为子宫内膜间质肿瘤。以往认为此类肿瘤为良性,最近的研究发现患者可发生转移和死亡,ICD编码为1,提示UTROSCT为交界性或生物行为不确定的肿瘤。根据NCOA1-3的伴侣基因不同,分为以下3种亚型。
3.1 伴ESR1重排的UTROSCT最常见的UTROSCT融合基因为ESR1∷NCOA3,好发于绝经前妇女,中位年龄47岁,中国患者发病中位年龄35岁[16-19]。此类肿瘤大部分位于黏膜下(图6A),少数位于肌壁间,罕见复发,预后好。肿瘤具有明显的性索样分化,如Sertoli样小管、网状结构(图6B)和条索状结构,有时可见上皮样细胞或簇状泡沫样细胞(图6C)。细胞异型性不明显,核分裂象罕见。目前,文献报道22例患者中仅1例于术后56个月发生盆腔复发,该例除经典的性索样形态外(图6D),还伴弥漫上皮样分化,但上皮样形态与预后关系需更多的数据加以证实。
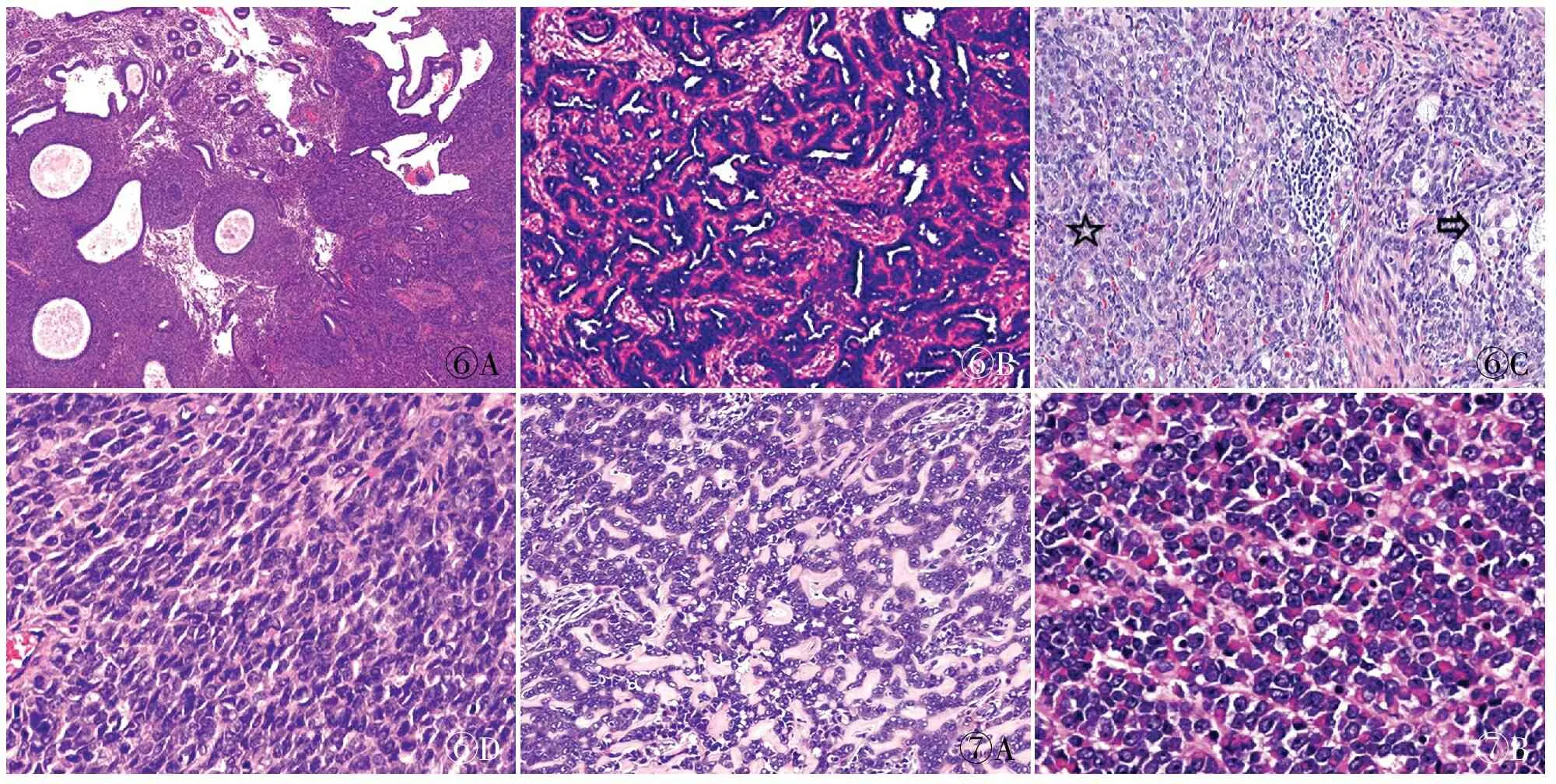
⑥A⑥B⑥C⑥D⑦A⑦B
UTROSCT中少见的融合基因为ESR1∷NCOA2,患者中位年龄40岁[16-17,20],镜下均伴性索样结构(图7A),近一半病例可见横纹肌样分化(图7B)。80%(4/5)伴横纹肌样分化的患者复发,NCOA2基因调节骨骼肌的生长,可形成横纹肌样细胞形态,提示可能具有侵袭性的生物学行为。
3.2 伴GREB1重排的UTROSCT与伴ESR1重排相比,伴GREB1重排的UTROSCT分化较差,患者发病年龄较大,好发于绝经后女性,中位年龄65岁,我国患者中位年龄48岁[17,19]。伴GREB1重排的UTROSCT肿瘤体积比ESR1重排更大,平均最大径7.0 cm,多数位于肌壁间,形态学多样且多为非特异性,有时性索形态不明显或局灶分布,导致诊断率低。肿瘤呈弥漫束状、巢状、漩涡状或偶见网状排列,也可见条索状结构伴间质胶原化或水肿(图8A~D),细胞呈梭形,细胞核圆形或卵圆形,核仁小而明显,核分裂象多少不等,比非GREB1重排的病例多见,可见坏死和脉管侵犯。伴GREB1重排的UTROSCT生物学行为更具侵袭性,可发生肺转移、盆腔复发导致死亡。
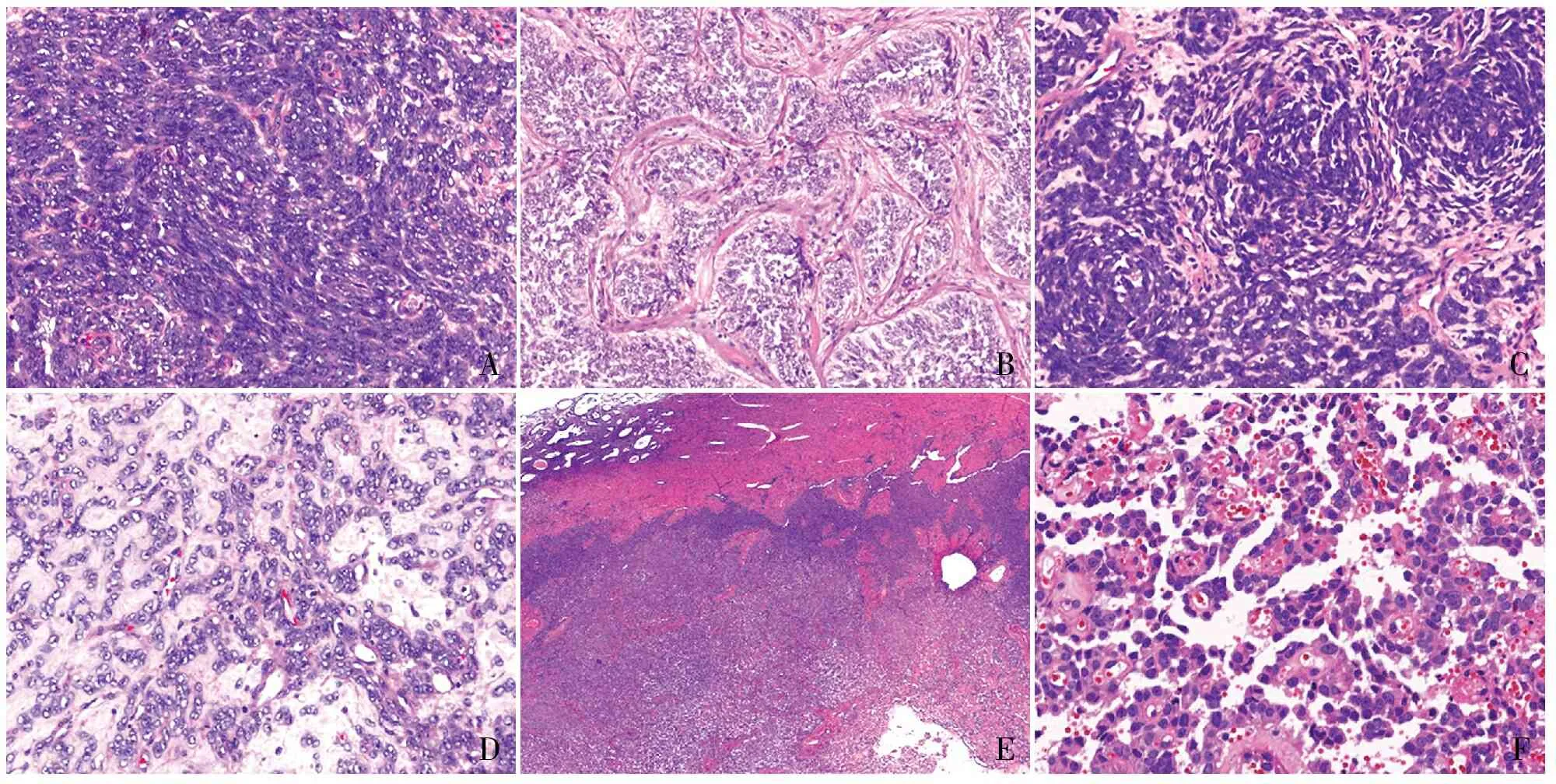
ABCDEF
目前,GREB1∷NCOA2是UTROSCT中复发率(57%)最高的融合基因,可见淋巴结转移,患者通常在2~14年复发或死亡[16-17,21]。复发灶与原发病灶形态相似,核分裂象可轻微增加或无明显变化。
GREB1∷NCOA1的UTROSCT肿瘤位于肌壁间(图8E),性索结构不明显,是误诊率最高的亚型。部分病例可出现肿瘤细胞围绕血管生长的假乳头状结构(图8F),易误诊为浆液性癌。多向分化的免疫表型和特征的分子改变有助于诊断。既往文献报道的病例均未复发,2023年Bi等[17]报道有2例盆腔复发,其中1例患者拒绝术后化疗,于44个月后死亡。
3.3 伴GTF2A1∷NCOA2的UTROSCT目前,文献仅报道2例伴GTF2A1∷NCOA2的UTROSCT[17,22]。镜下主要特征包括细胞密集的实性区与黏液样变的细胞稀疏区混杂存在,性索样结构不明显。实性区细胞呈梭形,细胞核中度异型,可见散在的伴奇异核细胞;在黏液样基质区可见上皮样细胞,胞质丰富嗜酸,单个或多个奇异性核,偶见核仁,核分裂少见,可见局灶坏死。1例于术后6个月复发,提示有侵袭性生物学行为(表2)。
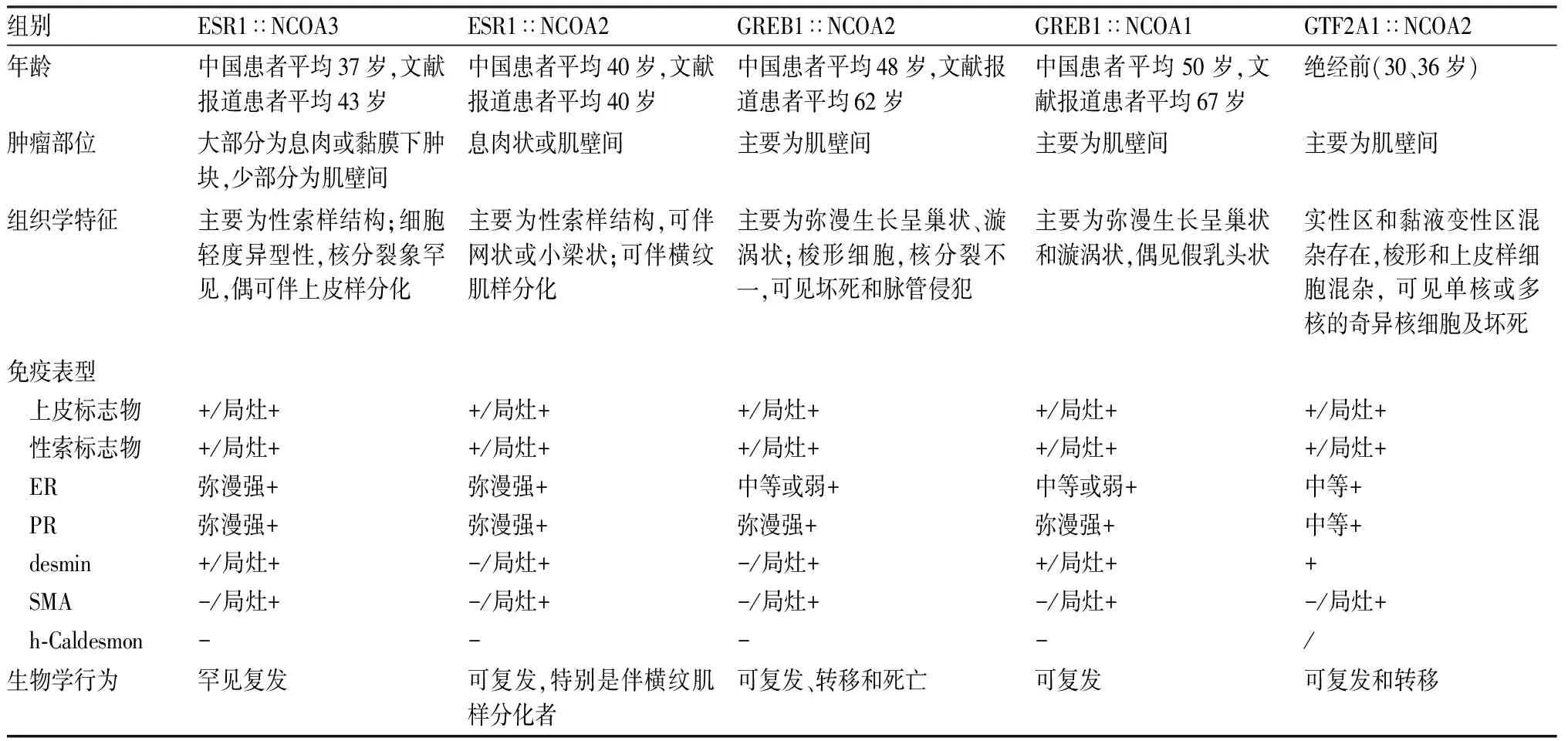
表2 不同分子亚型UTROSCT临床病理特征比较
4 新近出现的肿瘤类型
4.1 NTRK重排的子宫肉瘤NTRK家族基因包括NTRK1、NTRK2 和NTRK3。NTRK重排的梭形细胞肉瘤是以基因变异类型命名的肿瘤,第五版WHO分类将其单独列出。 NTRK重排的子宫肉瘤好发于绝经前女性,中位年龄39岁,发病部位多为子宫颈,少见于子宫体[23-25]。镜下肿瘤呈浸润性边界,梭形肿瘤细胞呈束状、鱼骨样杂乱排列(图9A),细胞间可见多少不等胶原纤维(图9B);细胞轻至中度异型,核卵圆形,染色质淡染,核仁小不明显,细胞质嗜酸(图9C);核分裂象多少不等(1~50个/10 HPF),与细胞异型性无关,偶可见非典型核分裂象和坏死。有时可见局灶黏液样基质,血管外皮瘤样生长,明显的淋巴细胞浸润,脉管侵犯通常不易见。免疫表型:pan-TRK阳性(图9D),因融合伴侣基因不同,染色定位不同;可不同程度地表达S-100、CD34、Cyclin D1和SMA,但不表达desmin、CD10、ER和PR等。分子检测发现大多数NTRK重排的子宫肉瘤主要融合形式为TPM3∷NTRK1,其他融合伴侣包括LMNA、TPR、RBPMS和EML4等[23-24]。目前,NTRK重排肉瘤的预后数据较少,有文献报道约1/3病例有复发或转移。Costigan等[25]报道与预后不良相关的特征包括:核分裂≥8个/10 HPF、淋巴管血管侵犯、坏死和伴NTRK3基因融合。NTRK重排肉瘤患者可在Trk抑制剂治疗中获益。

⑨A⑨B⑨C⑨D⑩A⑩B
4.2 COL1A1∷PDGFB基因融合的子宫肉瘤其临床罕见,与隆突性皮肤纤维肉瘤有形态学重叠,两者具有相同的融合基因和相似的免疫表型。COL1A1∷PDGFB基因融合的子宫肉瘤发病部位和组织学与NTRK重排肉瘤相似,梭形肿瘤细胞呈编织状(图10A),轻-中度细胞核异型,胞质稀少、嗜酸性,细胞边界不清,核分裂象易见(图10B);无脉管侵犯和淋巴细胞浸润[23]。患者发病年龄比NTRK重排肉瘤患者大,平均年龄58岁,具有低度恶性。该肿瘤表达CD34 和PDGFB,不表达pan-TRK、S-100、ER 和PR,有助于与NTRK重排肉瘤鉴别。伴PDGFB重排的子宫肉瘤患者,可从酪氨酸激酶抑制剂-伊马替尼的靶向治疗中获益[7]。
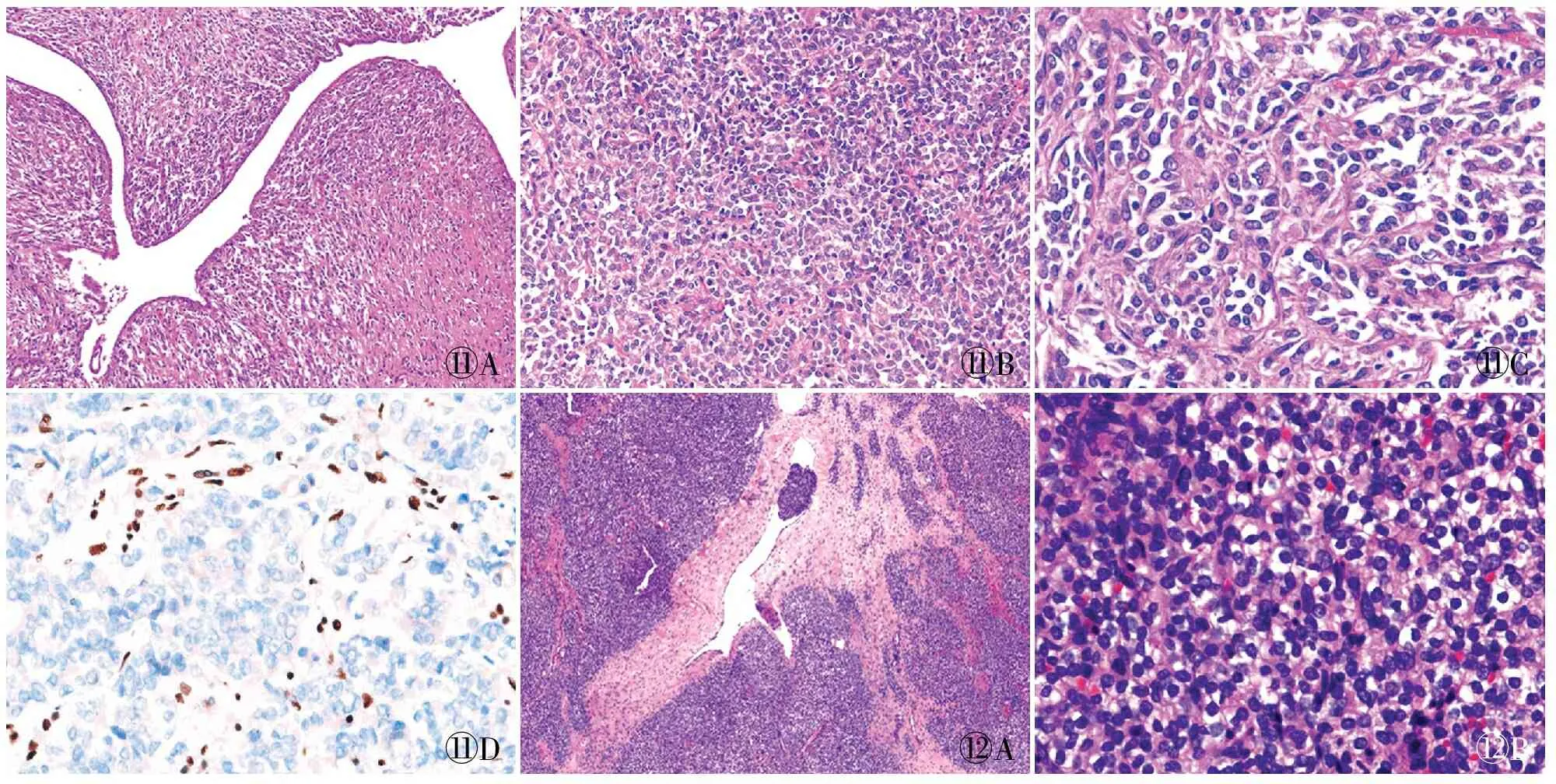
4.3 SMARCA4缺失的子宫未分化肉瘤(SMARCA4-deficient undifferentiated uterine sarcoma, SDUS)Kolin等[26]报道SDUS好发于年轻女性,中位年龄33岁。肿块侵犯子宫肌层,并累及附件、盆腔和淋巴结,患者预后差,中位生存期7~9个月。镜下可见良性的子宫内膜腺体内陷于肿瘤中,似叶状结构(图11A);主要为形态单一且黏附性差的上皮样肿瘤细胞呈片状分布 (图11B),细胞中等-大,胞质丰富嗜酸,泡状核,核仁明显(图11C),可伴明显的横纹肌样分化;部分病例可见小细胞或梭形细胞形态,模糊的巢状结构及局灶间质黏液样变性。核分裂象较活跃,脉管侵犯和坏死常见。此类肿瘤形态学与未分化和去分化子宫内膜癌(undifferentiated and dedifferentiated endometrial carcinomas, UDEC)重叠,后者也可伴SMARCA4缺失,但SDUS预后较UDEC更差。叶状结构更多见于SDUS,显著的核多形性更常见于UDEC。免疫组化染色有助于鉴别:SDUS常表现SMARCA4缺失(图11D),可伴SMARCA2缺失,上皮、平滑肌标记均阴性。此外,MMR缺失,p53、CK18、Claudin-4异常表达更多见于UDEC,而未在SDUS中表达。分子检测显示:SDUS以SMARCA4基因失活突变为特征,且均为微卫星稳定型[26],UDEC多伴子宫内膜癌相关的基因突变(如TP53、PTEN、PIK3CA)和微卫星不稳定。文献报道SDUS中有SMARCA4胚系突变者,伴发非典型横纹肌样瘤和卵巢高钙血症型小细胞癌的风险高。因此,识别该类患者对家族遗传学筛查很重要[27]。已有临床试验证实SWI/SNF复合体突变驱动的肿瘤中,PD-L1、EZH2和CDK4/6抑制剂等靶向治疗有重要价值[28-29]。这些治疗策略可能用于SDUS的治疗,值得进一步探索。
ABCDAB
4.4 伴GLI-1基因重排的间叶性肿瘤近年文献报道,伴GLI-1基因重排的肿瘤可发生于子宫[30- 31]。镜下肿瘤呈多结节状生长(图 12A),瘤细胞呈卵圆形和短梭形,巢状排列,肿瘤巢可见丰富的血管网 (图12B);部分间质可见胶原化或黏液样背景;可见坏死、核分裂象和脉管侵犯。不同部位的伴GLI-1基因重排的肿瘤免疫表型可不一致,多表达D2-40、S-100、CD10和Cyclin D1[30,32]。发生于子宫的伴GLI-1基因重排肿瘤需与LGESS或HGESS鉴别,分子检测有助于诊断。伴GLI-1基因改变的间叶源性肿瘤生物学行为和预后尚存争议。多数病例显示预后良好,但也有局部复发、淋巴结和远处转移导致死亡的报道[30-31]。靶向GLI-1的抑制剂已获得美国食品药品监督管理局(FDA)批准,有望为该类患者提供靶向治疗的机会。
5 小结
综上所述,随着子宫间叶源性肿瘤中具有特殊基因改变的新亚型不断发现,其临床病理特征逐渐清晰,有助于对新亚型的认识与诊断。挖掘子宫间叶源性肿瘤的分子特征,为精准诊断和靶向治疗提供更多的证据。病理医师应充分结合形态学特点和辅助检测结果,进行更加准确的诊断与鉴别诊断。
猜你喜欢
首都食品与医药(2023年14期)2023-07-17
中国药学药品知识仓库(2022年8期)2022-05-09
大学化学(2021年7期)2021-08-29
通信技术(2019年8期)2019-09-03
国际呼吸杂志(2019年4期)2019-03-12
医药前沿(2018年13期)2018-04-20
西安建筑科技大学学报(自然科学版)(2016年5期)2016-11-10
实用手外科杂志(2015年3期)2015-08-27
浙江理工大学学报(自然科学版)(2015年7期)2015-03-01
中国实用医药(2013年12期)2013-02-02
