
叠氮化银起爆药连续化合成芯片的设计与应用研究
2024-03-27 07:44韩瑞山王燕兰卢飞朋褚恩义
含能材料 2024年3期
韩瑞山,王燕兰,卢飞朋,张 松,张 方,李 蛟,褚恩义
(陕西应用物理化学研究所, 瞬态化学效应与控制全国重点实验室, 陕西 西安 710061)
0 引 言
叠氮化银(AgN3,简写为SA)是叠氮酸的银盐,最早在1890 年由Curtius 将叠氮酸(HN3)气体通入硝酸银溶液中制得。相较于目前广泛应用的叠氮化铅(LA)起爆药,SA 起爆药在水溶液中的溶解度更小,在制备时易大量、快速析出,不易产生针状大晶体,故而无自爆风险。同时,SA 起爆药具有更高的热稳定性、安定性,在真空环境下加热到400 ℃都不会发生爆炸[1-3],且干燥的SA 起爆药HN3分压非常低,与金属铜接触时基本不会反应生成高敏感的叠氮化铜[4]。另外,SA 起爆药的起爆能力高于LA,具有非常小的临界起爆尺寸[5-7],因此特别适合用于MEMS 火工品等小尺寸、低极限药量火工品装药。
SA 起爆药的制备大多使用AgNO3和NaN3的沉淀反应[8-9],对于沉淀反应而言,反应溶液混合速度的快慢,对于反应过程和结果都会产生重要影响[10-12]。微流控技术中的微混合芯片具备通道尺寸微小、反应参数精确可控、高效传质传热的特点[13-15],成为微纳米级叠氮化物起爆药制备的一种有效手段[16-18]。但微混合芯片中,反应液体的混合主要依靠通道壁面结构对流体产生折叠、扰动、分散或旋转等作用[19-22],混合效率受通道几何结构尺寸的影响极大。因此,设计优化微混合芯片通道结构,对于叠氮化物起爆药的连续化、高质量制备具有重要意义。
本研究设计并制作了一种连续反向旋T 形微混合芯片,采用Ansys Fluent 仿真模拟软件研究了芯片结构和流动条件等因素对芯片混合效率的影响,获得了最佳的芯片形状及其结构参数。并使用该芯片进行了SA 起爆药的连续化合成,分析了反应物流速、浓度和表面活性剂等因素对SA 起爆药粒径调控的影响,同时采用扫描电子显微镜(SEM)、粉末X 射线衍射(PXRD)、差示扫描量热(DSC)等手段对SA起爆药产物的形貌、成分、结构和热性能进行了表征和测试。
1 微混合芯片设计及优化
1.1 芯片结构设计及仿真
1.1.1 微混合芯片设计
T 形微混合通道是微混合芯片中研究最多的结构之一,其具有结构简单、流阻小等特点,但通道内流体基本为层流状态,混合效果受扩散系数影响较大,混合效率较低。通过将T 形微混合器中与主通道连接的两个侧通道入口设计一定的偏移量,使经过两入口流体在主通道内汇合时产生相反的速度矢量,呈现出旋涡状运动。随后在混合区,设置多个环形区,将混合溶液再次分离为两股液体,以相反的入口偏移量,重新汇入主通道,形成与上一个混合结构方向相反的涡流,多次重复以实现液体的高效混合。通道结构如图1 所示,其中d为通道尺寸,θ为两相溶液对撞角度,L为混合区域长度;P1,P2,P3,P4,P5 分别为混合效率计算截面位置,位于混合区域长度的中心。

图1 旋T 形微混合芯片通道结构示意图Fig.1 Schematic diagram of the channel structure in the rotating T-shaped micro-mixing chip
1.1.2 混合效率仿真模拟
使用Ansys Fluent 仿真模拟软件中多组分输运模型,求解各组分在整个微通道中的混合情况[15]。两相流体材料分别为NaN3和AgNO3的低浓度盐溶液,其密度都设为1020 kg·m-3,黏度为9.5×10-4kg·m-1·s-1,质量扩散系数为1.837×10-9m2·s-1。其中壁面条件设置为固定无滑移模式,入口条件设置为速度入口,速度大小按照实验时采用的流量数据换算后设置,出口条件设置为压力出口,相对压力为0。为方便观察和比较,设置模拟计算中入口处待混合流体质量分数分别为1 和0。微混合芯片的混合效率通过提取建立模型中微通道各截面上的溶液质量分数进行计算,如式(1)所示。
其中,M为混合效率,N为取点的个数,Xi为第i个点的质量分数,Xi-unmix为未混合情况下各点的质量分数,Xˉ为完全混合情况下各点质量分数的平均值。可以通过M值的大小来判断微混合芯片在不同截面处混合效率的高低,当溶液完全未混合时M= 0,当溶液完全混合时M= 100%。
1.2 仿真结果与讨论
1.2.1 混合通道尺寸对混合结果影响
为研究混合通道尺寸对旋T 形微混合芯片混合效率的影响,通过Fluent 仿真模拟了通道尺寸d分别为2.0,1.0,0.5 mm 的3 种芯片的混合效果,获得了流速为2 mL·min-1条件下3 种混合芯片的流线图及浓度压力分布云图。选取通道尺寸1.0 mm 的微混合芯片为代表,分析了增强混合原理,其内部两相溶液的流线图如图2 所示。可以看出,由于微通道交汇处存在一定的高度差,当两相溶液交汇时,产生不对称碰撞,溶液在主通道中发生旋流运动,促使通道内溶液发生旋转、折叠,从而增大了两相溶液间接触面积,缩短分子间扩散距离,促进了两相溶液的混合。随后混合溶液被再次分离为两股液体,并以相反的高度差重新交汇对撞,使其在主通道中形成与上一个混合结构方向相反的涡流,进一步增强混合。

图2 微混合芯片通道中两相溶液的流线图Fig.2 Streamline diagram of the two-phase solution in the micro-mixing chip channel
图3 为3 种尺寸微混合芯片在不同位置的浓度分布云图及混合效率,通过提取通道不同位置截面上的浓度分布数据,计算了不同位置处的混合效率。从图3 中可以看出,在相同截面位置处,随着微混合芯片通道尺寸减小,混合效率明显提升。这是由于随着通道尺寸减小,流体扩散距离也相应缩短,使得流体扩散混合速度加快。其中当混合芯片通道直径为2.0 mm 时,混合效率随截面位置的增加而缓慢增大;当混合芯片通道直径为1.0 mm 时,混合效率随截面位置的增加先迅速升高,后缓慢升高;而当混合芯片通道直径为0.5 mm 时,混合效率随截面位置先迅速升高至95%以上,后维持恒定。这是由于相同流量下,随着通道直径增大,通道中液体流动速度减小,通道交汇处碰撞作用减弱,流体混合基本依靠分子扩散完成,使得大通道直径中流体混合效率随截面位置增长缓慢。这说明不同通道尺寸和流速下,流体实现完全混合所需要的混合长度并不相同。
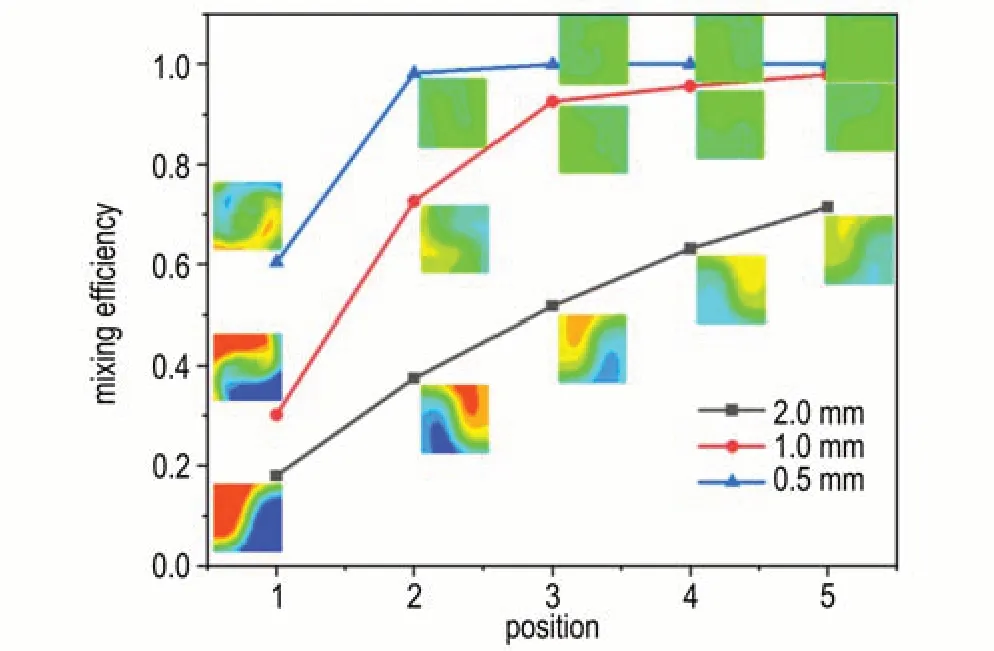
图3 3 种尺寸微混合芯片在不同位置的浓度分布云图及混合效率Fig.3 Concentration distribution contour and mixing efficiency of micro-mixing chips with three different sizes at various positions
图4 为不同通道尺寸的压力云图,可见随着通道尺寸的减小,3 种微混合芯片的最大压力升高值分别为33.5,297,2978 Pa,表明随着通道尺寸的减小,通道内流动阻力显著增大,但较大的流动阻力需要芯片及其接口具有很高的承压能力,不利于芯片及其接口的设计和制造。综合考虑通道流体阻力和混合效率,选用1.0 mm 通道尺寸的微混合芯片作进一步研究。

图4 3 种尺寸微混合芯片的压力分布云图Fig.4 Pressure distribution contour of three sizes of micro-mixing chips
1.2.2 对撞角度对混合结果影响
为研究两相溶液对撞角度θ对混合效率的影响,设计了对撞角度分别为120°和60°的微混合芯片,并在4 mL·min-1流速下,与180°微混合芯片进行了对比。3 种对撞角度的微混合芯片在不同截面上的混合效果图如图5 所示,可以看出,随着截面位置的增加,3 种对撞角度的微混合芯片混合效率都发生了显著升高,其中180°对撞的微混合芯片具有最高的混合效率。随着对撞角度的减小,在P1 截面上的混合效率随着对撞角度的降低而降低。但在P2 截面以后,60°对撞微混合芯片的混合效率反而大于120°微混合芯片。这表明,微混合芯片的混合效率并不随对撞角度单调变化,而是存在先降低后升高的过程。
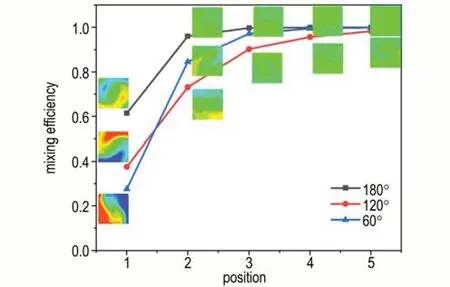
图5 3 种对撞角度微混合芯片在不同位置的混合效率及浓度分布云图Fig.5 Concentration distribution contour and mixing efficiency of three types of micro-mixing chips with different collision angles at various positions
1.2.3 反应物流速对混合结果影响
为研究反应物流速Q对混合效果的影响,针对通道直径1.0 mm 的微混合芯片,模拟计算了在流速分别为1,2,4,8 mL·min-1条件下的混合效果。4 种流速下微混合芯片在不同截面位置的浓度分布云图及混合效率,如图6 所示。可以看出,1 mL·min-1流速下微混合芯片混合效率较差,在经过P5 截面时只有70%的混合效率。随着溶液流速的增加,混合效率迅速升高。在2 mL·min-1流速下,反应溶液在流经微混合芯片的第3 个截面时就可达到90%以上的混合效率,流经P5 截面时达到97%以上的混合效率。而当流速为4 mL·min-1或8 mL·min-1时,反应溶液在流经微混合芯片的P3 截面时就可实现近100%的混合。这是由于较高流速容易在通道中形成更明显的涡旋结构,带来更大的扰动,促使混合效率升高。同时8 mL·min-1流速下溶液的混合效率与4 mL·min-1时基本相当,表明反应物流速≥4 mL·min-1时微混合芯片的混合效率已达到最优。
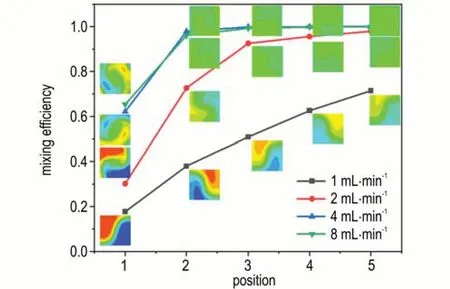
图6 4 种流速下微混合芯片在不同位置的混合效率及浓度分布云图Fig.6 Concentration distribution contour and mixing efficiency of micro-mixing chips at different positions under four flow rates
2 实验部分
2.1 试剂与仪器
试剂:AgNO3,分析纯,成都科隆化学品有限公司;NaN3,分析纯,郑州派尼化学试剂厂;十二烷基硫酸钠(SDS),分析纯,上海国药集团;十二烷基酚聚氧乙烯醚-10(OP-10),一级试剂,国药集团化学试剂有限公司;聚乙烯醇(PVA),PVA-1788,天津科密欧化学试剂有限公司。
仪器:VEGA TS5136XM 型扫描电子显微镜,捷克泰斯肯公司;D8 advance 型X 射线衍射仪,德国布鲁克公司;Nicolet 6700 型傅里叶变换红外光谱仪,美国赛默飞世尔科技公司;DSC204F1 型差示扫描量热仪,德国耐驰公司;TS-1B恒流泵,中国保定兰格恒流泵有限公司。旋T形微混合芯片,苏州汶灏微流控技术有限公司,有机玻璃材质,芯片通道深度0.5~1 mm,宽度1 mm。
2.2 SA 起爆药制备及表征
2.2.1 SA 起爆药制备
SA 起爆药的快速沉淀反应按以下反应方程式进行:
微流控技术合成SA 起爆药:将一定量AgNO3、NaN3分别溶解于去离子水中,按实验要求加入不同的表面活性剂,配制出浓度分别为0.01,0.02,0.03 mol·L-1的AgNO3和NaN3溶液。采用多通道恒流泵分别控制反应物流速,将两种溶液输送到旋T 型微流控芯片中,通过在旋T 形微混合芯片快速混合,反应生成AgN3。收集从旋T 型微流控芯片出口流出的沉淀产物,经多次离心和洗涤收集沉淀,在水浴烘箱内55 ℃干燥。实验装置示意图如图7 所示。

图7 旋T 型微流控芯片中SA 起爆药合成装置示意图Fig.7 Schematic representation of the synthesis apparatus for SA primary explosive
常规批次合成SA 起爆药:参照文献[23]的方法,室温下,配制50 mL 浓度为0.02 mol·L-1的AgNO3溶液作为底液,将相同浓度的NaN3溶液滴加至AgNO3溶液中,搅拌反应1 h,将反应溶液过滤、洗涤、烘干后,得到产物。
2.2.2 SA 起爆药的表征测试
采用扫描电子显微镜对不同条件制备的SA 起爆药颗粒形貌进行表征,样品均匀分散在导电胶上,经喷金后测试。
SA 起爆药的粒径及其分布依据GJB5891.6-2006方法对扫描电镜数据中晶体颗粒的投影面积逐个测量记录,以等效圆直径为粒径数据进行统计后计算分析得到样品的粒径分布[24]。
采用PXRD 分析SA起爆药颗粒的物相组成,分辨率为0.02°,扫描范围为5°~70°,扫描速率为10°·min-1。
采用FTIR 对SA 起爆药颗粒的分子结构进行表征,采用KBr 压片法,在400~4000 cm-1波数范围内测试,分辨率为4 cm-1。
采用DSC 进行热量测定试验,使用铟标准样品校准熔融温度和焓。分别将重量约为0.3 mg 的微流SA和常规SA 起爆药粉末样品放入到铝坩埚中,在氮气氛围中进行DSC 分析,氮气流速50 mL·min-1,样品测试温度范围为25~450 ℃,升温速率为10 ℃·min-1。
2.3 制备工艺对SA 形貌的影响
2.3.1 反应物流速对SA 合成影响
为考察反应物溶液在微通道中的流速Q对沉淀产物粒径的影响,使用浓度为0.02 mol·L-1的AgNO3及NaN3溶液,通过恒流泵调整反应溶液的流动速度,依据恒流泵量程,设置流速分别为8,4,2 mL·min-1。图8a~8c 为不同流速时制备的SA 起爆药晶体形貌。可以看出,所制备的SA 起爆药晶粒呈柱状或棒状,晶粒尺寸分布在0.38~2.45 μm 的范围内。随着流速减小,SA 起爆药的粒径及其分布明显增大。其中当反应流速为8 mL·min-1时,SA 起爆药的平均粒径最小,为0.77 µm(图8a);当反应流速降低为4 mL·min-1时,SA 起爆药的平均粒径增大为1.14 µm(图8b);当流速降为2 mL·min-1时,SA 起爆药具有最大的平均粒径,为1.24 µm(图8c),且粒径分布也最宽。这是由于旋T 形微混合芯片在较低的流速下混合效率较差,反应溶液不能很快地完成混合。
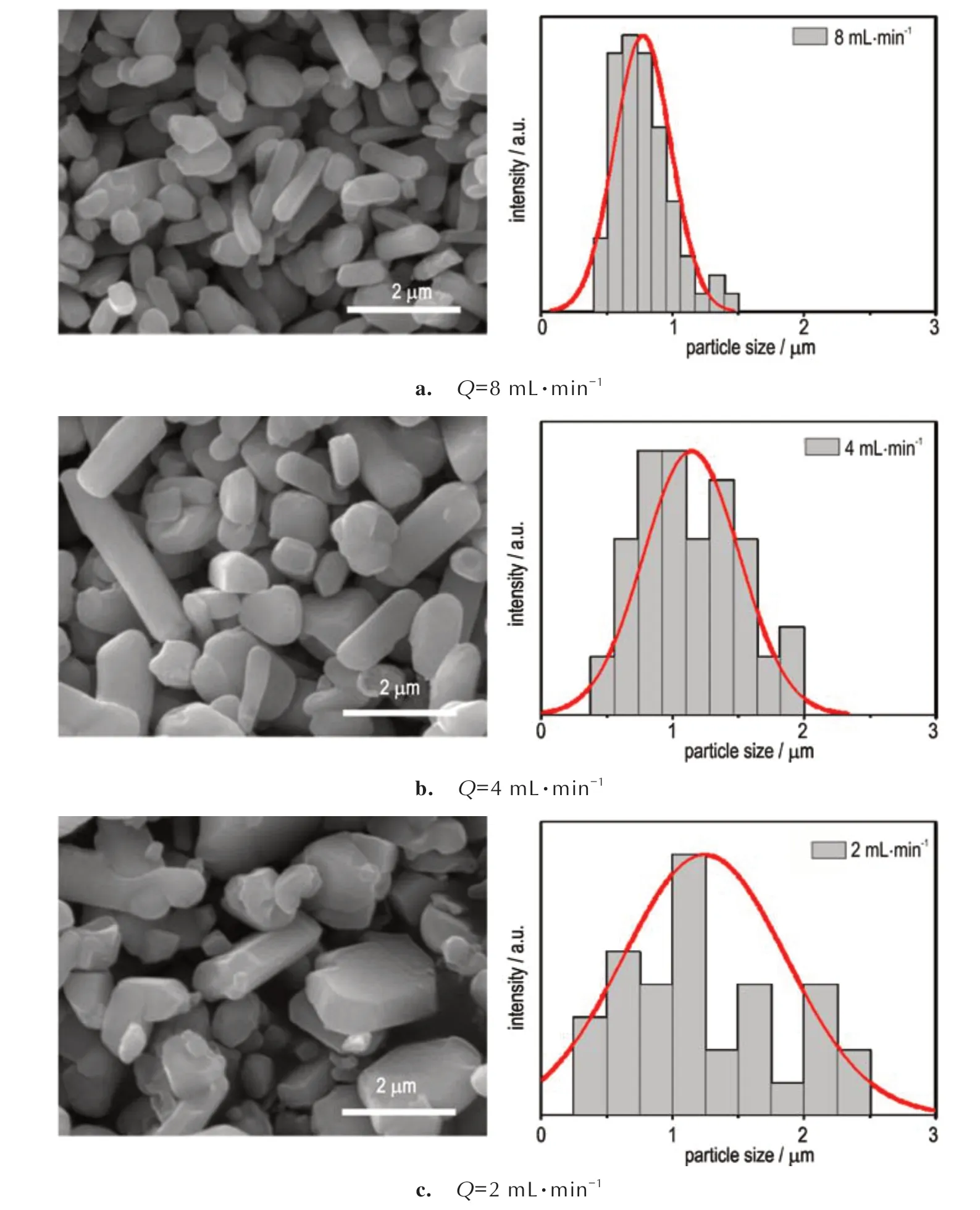
图8 不同流速下制备的SA 起爆药SEM 照片及粒径分布图Fig.8 SEM and particle size distribution images of SA primary prepared under different flow rates
2.3.2 反应物浓度对SA 合成影响
为考察反应物溶液浓度c对沉淀产物形貌的影响,分别配制了浓度为0.01,0.02,0.03 mol·L-1的AgNO3、NaN3溶液,使用4 mL·min-1反应流速进行了SA 起爆药的制备,其产物SEM 照片和粒径分布分别如图9a~9c所示。可以看出,当反应物浓度为0.01 mol·L-1时,沉淀产物为块状颗粒,粒径分布范围为0.68~2.14 μm,平均粒径为1.24 μm(图9a);反应物浓度为0.02 mol·L-1时,粒径分布范围为0.39~1.71 μm,平均粒径为0.79 μm(图9b);反应物浓度为0.03 mol·L-1时,粒径分布范围为0.22~1.26 μm,平均粒径为0.52 μm(图9c)。表明随着反应物浓度的增大,反应产物的粒径逐渐减小,且粒度分布范围也相应变窄。其原因是在一定的过饱和度条件下,晶核只有大于临界尺寸时才能稳定的存在,进而自发生长。随着反应物浓度增大,其结晶驱动力增大,临界成核半径减小,使得反应能够生成更多的晶核,致使产物粒径减小。
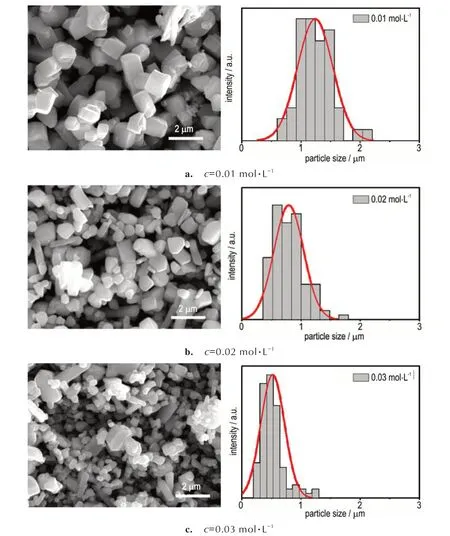
图9 反应物浓度对SA 起爆药粒径的影响Fig.9 Influence of reactant concentration on the particle size of SA primary explosive
2.3.3 表面活性剂对SA 合成影响
表面活性剂也会对沉淀结晶过程产生影响。为研究不同类型表面活性剂对SA 结晶过程的影响规律,选用实验室中常见的非离子型(OP-10)、聚合物型(PVA)和 阴 离 子 型(SDS)表 面 活 性 剂,添 加 于0.03 mol·L-1的NaN3溶液中,使用8 mL·min-1反应流速进行了SA 起爆药的制备,考察了表面活性剂对SA起爆药合成的影响。图10a~10d 分别为NaN3溶液中未添加表面活性剂和加入1%的OP-10、PVA、SDS 获得的SA 起爆药晶体SEM 照片和粒径分布图。可以看出,未加入表面活性剂的SA 起爆药颗粒基本为六棱柱形或球形,平均粒径为0.36 μm,粒径分布范围为0.14~0.60 μm(图10a)。而加入OP-10 和SDS 后,SA起爆药颗粒形貌呈片状形态,且产物粒径及分布范围显著增大,其中加入OP-10 的平均粒径为0.60 μm(图10b),加入SDS 的平均粒径为0.40 μm(图10d),这是由于OP-10 和SDS 改变了晶体上各个晶面上的相对生长速度,从而调控了结晶形态和粒径。需要特别说明的是,加入PVA 的SA 起爆药颗粒仍为六棱柱形或球形,平均粒径为0.07 μm,分布范围为0.03~0.21 μm(图10c),获得了比未填加表面活性剂时更小的颗粒尺寸和分布,这可能是由于PVA 降低了SA 晶核的形成能量,使得反应溶液能够快速生成大量的晶核,并生长为SA 晶粒。但由于PVA 为高分子型表面活性剂,在产物中不易去除且残留较多,因此后续SA制备过程中不进行表面活性剂的添加。
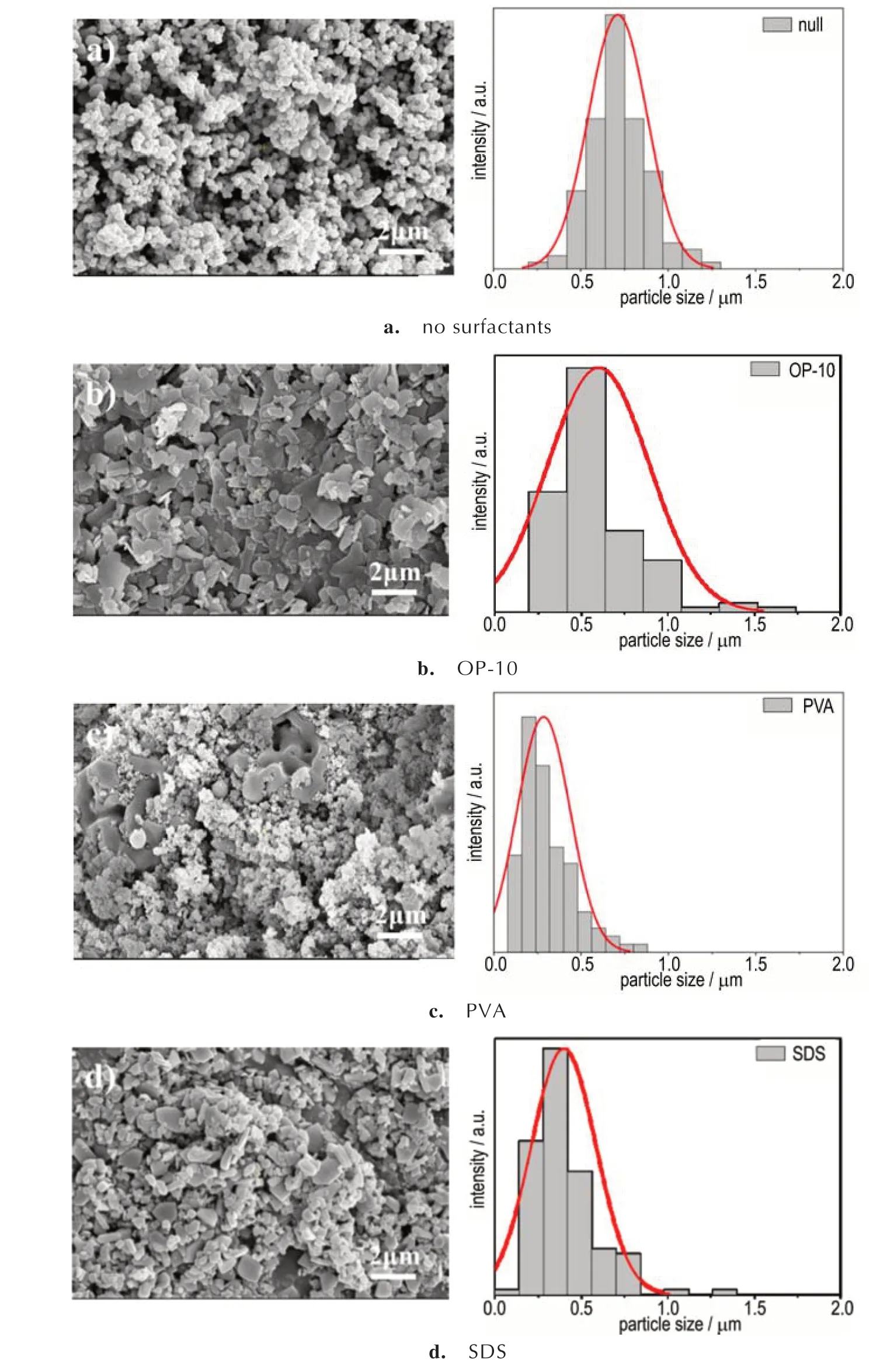
图10 表面活性剂对SA 起爆药粒径及其分布的影响Fig.10 Influence of surfactants on the particle size of SA primary explosive
2.4 SA 起爆药的测试结果分析
2.4.1 结构表征
为获得最优条件下微混合芯片制备的SA 起爆药的成分和晶体结构信息,使用溴化钾压片的方法进行了微流SA 的红外吸收测试,谱图如图11a 所示。可以看出,在2000~2200 cm-1处存在一个较强的吸收峰,这是基团中N=N=N 累积双键的伸缩振动峰,为叠氮化物起爆药的特征峰。1636 cm-1处的吸收峰是N=N(叠氮基)双键的伸缩振动峰,故而两者同时存在。而在3000~3600 cm-1处存在一个小的鼓包,这是由于样品中含有水造成的。同时在3355 cm-1和3298 cm-1两处存在尖锐吸收峰,推测是由于叠氮基团与样品中的水相互作用形成了H…N 氢键所引起的。
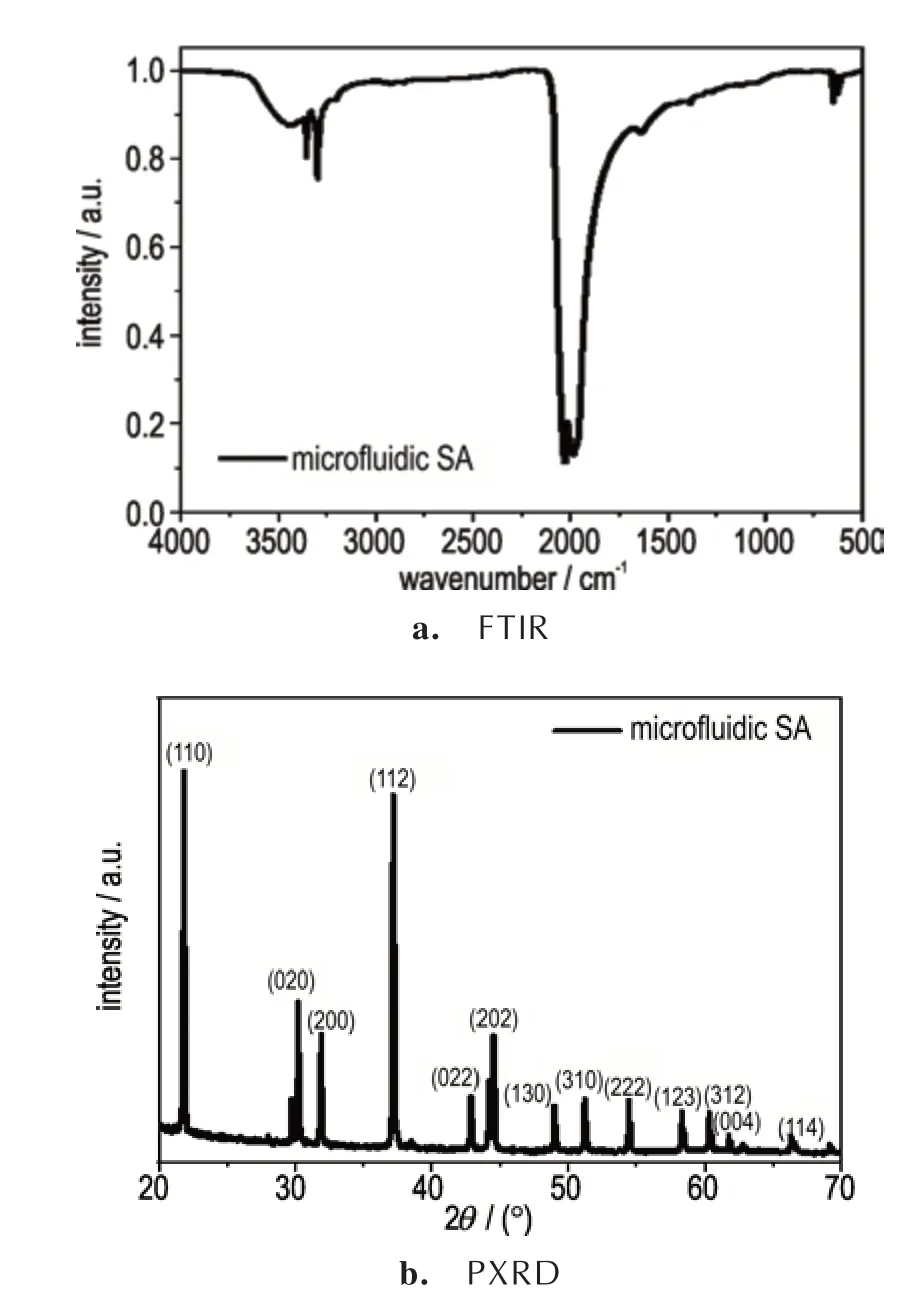
图11 微流SA 的FTIR 和PXRD 谱图Fig.11 FTIR and PXRD spectra of microfluidic SA
为进一步表征产物晶体结构,使用PXRD 对制备的SA 起爆药进行了测试,谱图如图11b 所示。可以看出,在21.84° ,30.23° ,31.90° ,37.19° ,42.93° ,44.49°,48.99°,51.22°,54.47°,58.37°和60.37°处有明显的衍射峰,分别对应SA 起爆药的(110),(020),(200),(112),(022),(202),(130),(310),(222),(123)和(312)晶面,制备的SA 起爆药主要成分都为正交晶系的AgN3晶体,晶面择优取向为(110)和(112)晶面。
2.4.2 热性能测试
为比较采用微流控方法制备的微流SA 起爆药,与常规批次反应法获得SA 起爆药之间的热性能差异,在10 ℃·min-1升温速率下分别对微流SA 起爆药和常规批次SA 起爆药进行了DSC 测试,谱图如图12 所示。由图12 可见,两种样品在306.5 ℃左右存在一个明显的吸热峰,这是由于SA 起爆药独特的物理化学性质所造成的,其在受热时存在先融化后分解的过程。与常规批次制备的SA 起爆药相比,微流SA 起爆药的放热峰温度由365.2 ℃降低到了358.2 ℃,降低了7 ℃,且放 热 量 由851.6 kJ·kg-1升 高 至976.7 kJ·kg-1,提 升 了14.7%,这表明使用微流控方法制备的SA 起爆药具有更高的反应活性和能量。

图12 微流SA 和常规批次SA 的DSC 曲线Fig.12 DSC curves of microfluidic SA and batch SA
3 结 论
设计并制作了一种连续反向旋T 形微混合芯片,研究了芯片结构及反应物流速对芯片混合效率的影响规律,并使用该芯片进行了SA 起爆药的连续化合成,探究了反应物流速、浓度和表面活性剂等因素对SA 起爆药粒径的影响,使用SEM、PXRD、DSC 等手段测试了样品的形貌、成分、结构和热性能,得到结论如下:
(1)旋T 形微混合芯片的混合效率与反应物流速密切相关。微混合芯片在通道尺寸为1 mm,对撞角度90°、流速4 mL·min-1以上时,可获得接近100%的混合效率;
(2)反应流速8 mL·min-1、浓度0.03 mol·L-1制备条件下可以获得形貌均匀、粒径分布较窄的类球形SA起爆药颗粒(平均粒径0.36 μm),且通过控制旋T 型微流混合芯片内反应物流速、反应物浓度、添加表面活性剂等条件,可实现对SA 起爆药晶形及粒度分布的有效调控;
(3)微流控方法制备的SA 起爆药相较常规批次SA 起爆药,放热峰温度提前了7 ℃(358.2 ℃ vs 365.2 ℃),放 热 量 升 高 了14.7%(851.6 kJ·kg-1vs 976.7 kJ·kg-1),说明其具有更高的反应活性和能量。
猜你喜欢
中学生数理化·八年级物理人教版(2023年4期)2023-05-05
中学生数理化·八年级物理人教版(2022年4期)2022-04-26
大众科学(2020年7期)2020-10-26
小天使·六年级语数英综合(2018年1期)2018-10-08
东华大学学报(自然科学版)(2018年1期)2018-06-29
中学化学(2017年5期)2017-07-07
中学化学(2016年4期)2016-05-30
分析测试学报(2015年4期)2016-01-13
分析测试学报(2015年3期)2016-01-13
分析测试学报(2015年3期)2016-01-13
